Illumina Run Processing Pipeline¶
Genomic Analyzers create massive amounts of data. The Illumina Run Processing Pipeline transforms primary imaging output from the Genome Analyzer into discrete aligned strings of bases. A package of integrated algorithms perform the core primary data transformation steps: image analysis, intensity scoring, base calling, and alignment. It also converts Base Call (BCL) files into FASTQ files that are needed for higher genomic analysis such as ChIP Sequencing.

Figure: Pipeline data transformation steps (Source: Illumina)¶
Introduction¶
The standard MUGQIC Illumina Run Processing pipeline uses the Illumina bcl2fastq software to convert and demultiplex the base call files (BCL) to FASTQ files. The pipeline runs some QCs on the raw data, on the FASTQ and on the alignment. It performs the core primary data transformation steps: image analysis, intensity scoring, base calling, and alignment.
Image Analysis: interpreting the image data to identify distinct clusters of genes
Base Calling: profiles for each cluster are used to call bases. Obtaining the quality of each base call is crucial for downstream analysis.
Alignment: entire set of called sequence reads are aligned to create a final sequence output file optimized for SNP identification.
Sample Files
This pipeline uses two input sample sheets.
Casava Sheet
Nanuq Run Sheet
You can see samples of Casava Sheet and Nanuq Run Sheet and also refer to Casava User Guide for more details.
Version¶
3.6.2
For the latest implementation and usage details refer to Illumina Sequencing implementation README file file.
Usage¶
illumina_run_processing.py [-h] [--help] [-c CONFIG [CONFIG ...]]
[-s STEPS] [-o OUTPUT_DIR]
[-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}]
[--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-d RUN_DIR] [--lane LANE_NUMBER]
[-r READSETS] [-i CASAVA_SHEET_FILE]
[-x FIRST_INDEX] [-y LAST_INDEX]
[-m NUMBER_OF_MISMATCHES] [-w] [-v]
Optional Arguments
-d RUN_DIR, --run RUN_DIR
run directory
--lane LANE_NUMBER lane number
-r READSETS, --readsets READSETS
nanuq readset file. The default file is
'run.nanuq.csv' in the output folder. Will be
automatically downloaded if not present.
-i CASAVA_SHEET_FILE illumina casava sheet. The default file is
'SampleSheet.nanuq.csv' in the output folder. Will be
automatically downloaded if not present
-x FIRST_INDEX first index base to use for demultiplexing
(inclusive). The index from the sample sheet will be
adjusted according to that value.
-y LAST_INDEX last index base to use for demultiplexing (inclusive)
-m NUMBER_OF_MISMATCHES
number of index mistmaches allowed for demultiplexing
(default 1). Barcode collisions are always checked.
-w, --force-download force the download of the samples sheets (default:
false)
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute Illumina Sequencing Pipeline:
illumina_run_processing.py <Add options - info not available in README file> >illumina_cmd.sh
bash illumina_cmd.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline here.
Pipeline Schema¶
Figure below shows the schema of the Illumina Sequencing workflow.
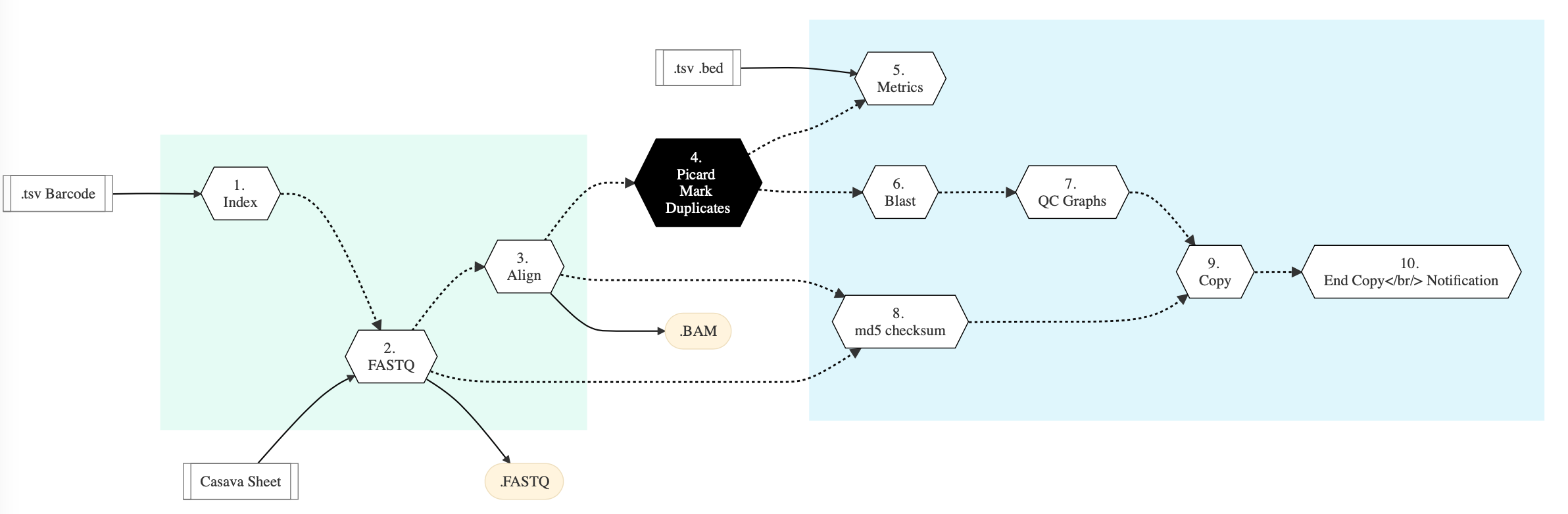
Figure: Schema of Illumina Sequencing protocol¶
Pipeline Steps¶
The table below shows various steps that are part of Illumina Sequencing Pipeline:
Illumina Sequencing Steps |
|
|---|---|
Step Details¶
Following are the various steps that are part of GenPipes Illumina Sequencing genomic analysis pipeline:
Index
This step generates a file with all the indexes found in the index-reads of the run.
The input barcode file is a two columns tsv file. Each line has a barcode_sequence and the corresponding barcode_name. This file can be generated by a LIMS.
The output is a tsv file named RUNFOLDER_LANENUMBER.metrics that will be saved in the output directory. This file has four columns, the barcode/index sequence, the index name, the number of reads and the number of reads that have passed the filter.
FASTQ Step
This step launches FASTQ generation from Illumina raw data using BCL2FASTQ conversion software. The index base mask is calculated according to the sample and run configuration; and also according the mask parameters received (first/last index bases). The Casava sample sheet is generated with this mask. The default number of mismatches allowed in the index sequence is 1 and can be overridden with an command line argument. No demultiplexing occurs when there is only one sample in the lane.
An optional notification command can be launched to notify the start of the fastq generation with the calculated mask.
Align
In this step, reads are aligned from the fastq file, sort the resulting .bam and create an index of that .bam. An basic alignment is performed on a sample when the SampleRef field of the Illumina sample sheet match one of the regexp in the configuration file and the corresponding genome (and indexes) are installed. STAR Software is used as a splice-junctions aware aligner when the sample library_source is cDNA or contains RNA; otherwise BWA_mem is used to align the reads.
For details, refer to STAR User Guide.
Picard Mark Duplicates
Runs Picard mark duplicates on the sorted BAM file.
Metrics
This step runs a series of multiple metrics collection jobs and the output bam from mark duplicates.
Picard CollectMultipleMetrics: A collection of picard metrics that runs at the same time to save on I/O. - CollectAlignmentSummaryMetrics, - CollectInsertSizeMetrics, - QualityScoreDistribution, - MeanQualityByCycle, - CollectBaseDistributionByCycle
BVATools DepthOfCoverage: Using the specified
BED Filesin the sample sheet, calculate the coverage of each target region.Picard CalculateHsMetrics: Calculates a set of Hybrid Selection specific metrics from the BAM file. The bait and interval list is automatically created from the specified BED Files.
BLAST Step
Runs BLAST on a subsample of the reads of each sample to find the 20 most frequent hits. The runBlast.sh tool from MUGQIC Tools is used. The number of reads to subsample can be configured by sample or for the whole lane. The output will be in the Blast_sample folder, under the Unaligned folder.
QC Graphs Step
Generate some QC Graphics and a summary XML file for each sample using BVATools.
Files are created in a ‘qc’ subfolder of the fastq directory. Examples of output graphic:
Per cycle qualities, sequence content and sequence length;
Known sequences (adapters);
Abundant Duplicates
MD5 Step
Create md5 checksum files for the fastq, BAM and BAI using the system ‘md5sum’ utility. One checksum file is created for each file.
Copy Step
Copy processed files to another place where they can be served or loaded into a LIMS. The destination folder and the command used can be set in the configuration file. An optional notification can be sent before the copy. The command used is in the configuration file.
End Copy Notification
Send an optional notification to notify that the copy is finished. The command used is in the configuration file. This step is skipped when no command is provided.