Nanopore Pipeline¶
Structural Variations (SV) are genomic alterations including insertions, deletions, duplications, inversions, and translocation. They account for approximately 1% of the differences among human genomes and play a significant role in phenotypic variation and disease susceptibility.
Nanopore sequencing technology can generate long sequence reads and provides more accurate SV identification in terms of both sequencing and data analysis. For SV analysis, several new aligners and SV callers have been developed to leverage the long-read sequencing data. Assembly based approaches can also be used for SV identification. Minimap2 aligner offers high speed and relatively balanced performance for calling both insertions as well as deletions.
The Nanopore sequencing technology commercialized by Oxford Nanopore Technologies (ONT).
Introduction¶
The Nanopore is used to analyze long reads produced by the Oxford Nanopore Technologies (ONT) sequencers. Currently, the pipeline uses Minimap2 to align reads to the reference genome. Additionally, it produces a QC report that includes an interactive dashboard for each readset with data from the basecalling summary file as well as the alignment. A step aligning random reads to the NCBI nucleotide database and reporting the species of the highest hits is also done as QC.
Once the QC and alignments have been produced, Picard is used to merge readsets coming from the same sample. Finally, SVIM is used to detect Structural Variants (SV) including deletions, insertions and translocations. For a full summary of the types of SVs detected, please consult the following site.
The SV calls produced by SVIM are saved as VCFs for each sample, which can then be used in downstream analyses. No filtering is performed on the SV calls.
This pipeline currently does not perform base calling and requires both FASTQ and a sequencing_summary file produced by a ONT supported basecaller (we recommend Guppy). Additionally, the testing and development of the pipeline were focused on genomics applications, and functionality has not been tested for transcriptomic or epigenomic datasets. Beyond the QC dashboards for each readset, there is currently no implemented reporting step in this pipeline.
For more information on using ONT data for structural variant detection, as well as an alternative approach, please consult Oxford Nanopore Technologies SV Pipeline GitHub repository.
For information on the structure and contents of the Nanopore readset file, please consult Nanopore Readsets details.
Version¶
3.6.2
For the latest implementation and usage details refer to Nanopore Sequencing implementation README file file.
Usage¶
nanopore.py [-h] [--help] [-c CONFIG [CONFIG ...]]
[-s STEPS] [-o OUTPUT_DIR]
[-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}]
[--sanity-check]
[--container {wrapper, singularity} <IMAGE FILE>
[--genpipes_file GENPIPES_FILE]
[-r READSETS] [-v]
Optional Arguments
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute Nanopore Sequencing Pipeline:
nanopore.py <Add options - info not available in README file -g nanopore_cmd.sh
bash nanopore_cmd.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline here.
Pipeline Schema¶
The following figure shows the schema for Nanopore sequencing pipeline:
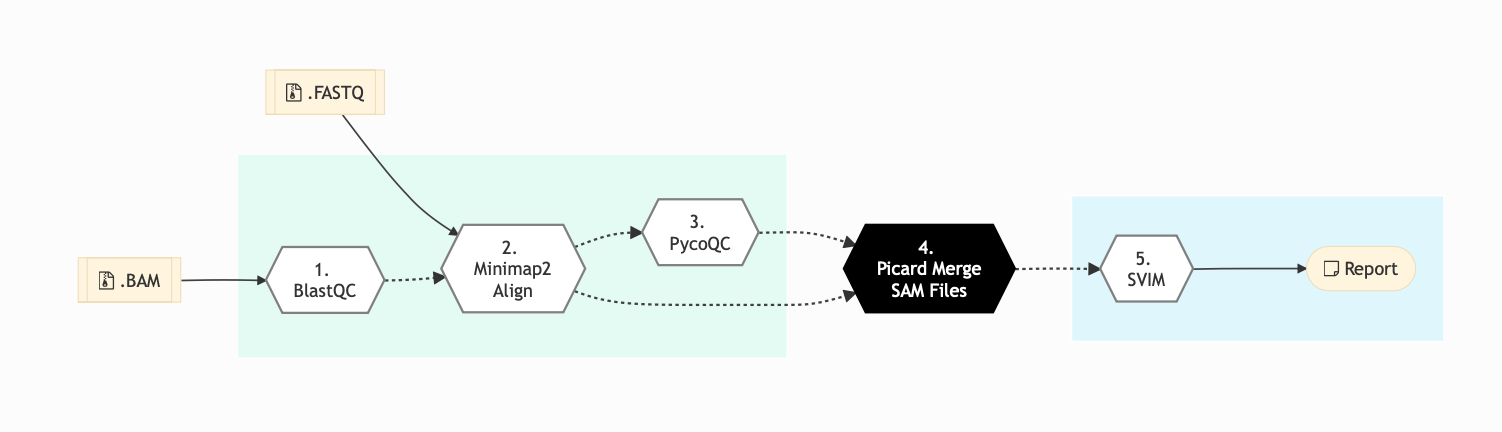
Figure: Schema of Nanopore Sequencing protocol¶
Pipeline Steps¶
The table below shows various steps that are part of Nanopore Sequencing Pipeline:
Nanopore Sequencing Steps |
|
|---|---|
Step Details¶
Following are the various steps that are part of GenPipes Nanopore genomic analysis pipeline:
BlastQC
In this step, Blast-QC utility is used for sequence alignment and identification. It performs a basic QC test by aligning 1000bp of randomly selected reads to the NCBI Nucleotide Database in order to detect potential contamination.
Minimap2 Align
Minimap2 Align Program is a fast, general purpose sequencing alignment program that maps DNA and long mRNA sequences against a large reference database. It can be used for Nanopore sequencing for mapping 1kb genomic reads at an error rate of 15% (e.g., PacBio or Oxford Nanopore genomic reads), among other uses.
In this step, minimap2 to align the Fastq reads that passed the minimum QC threshold to the provided reference genome. By default, it aligns to the human genome reference (GRCh38) with Minimap2.
pycoQC
In this step, pycoQC Software is used produce an interactive quality report based on the summary file and alignment outputs. PycoQC relies on the sequencing_summary.txt file generated by Guppy. If needed, it can also generate a summary file from basecalled FAST5 files. PycoQC computes metrics and generates interactive QC plots for Oxford Nanopore technologies sequencing data.
Picard Merge SAM Files
BAM readset files are merged into one file per sample in this step. Using aligned and sorted BAM output files from Minimap2 Align step, it performs the merge using Picard.
Structural Variant Identification using Mapped Long Reads
In this step, Structural Variant Identification using Mapped Long Reads (SVIM methodology), is used to perform structural variant (SV) analysis on each sample.
More Information¶
Evaluating nanopore sequencing data processing pipelines for structural variation identification.
Minimap2: Pairwise alignment for nucleotide sequences.