DNA Sequencing Pipeline¶
DNA sequencing is the process of determining the sequence of nucleotides in a section of DNA. Next-generation sequencing (NGS), also known as high-throughput sequencing, allows for sequencing of DNA and RNA much more quickly and cheaply than the previously used Sanger sequencing, and as such revolutionized the study of genomics and molecular biology. The enormous amount of short reads generated by the new DNA sequencing technologies call for the development of fast and accurate read alignment programs.
Over the past decade, long-read, single-molecule DNA sequencing technologies have emerged as powerful players in genomics. With the ability to generate reads tens to thousands of kilobases in length with an accuracy approaching that of short-read sequencing technologies, these platforms have proven their ability to resolve some of the most challenging regions of the human genome, detect previously inaccessible structural variants and generate some of the first telomere-to-telomere assemblies of whole chromosomes.
Burrows-Wheeler Alignment tool, BWA, is a new read alignment package that is based on backward search with Burrows–Wheeler Transform (BWT), to efficiently align short sequencing reads against a large reference sequence such as the human genome, allowing mismatches and gaps. BWA is ∼10–20 times faster than Mapping and Assembly with Quality, MAQ, while achieving similar accuracy. In addition, BWA outputs alignment in the new standard SAM (Sequence Alignment/Map) format. Variant calling and other downstream analyses after the alignment can be achieved with the open source SAMtools software package.
DNA Sequencing GenPipes pipeline has been implemented by optimizing the Genome Analysis Toolkit, GATK best practices standard operating protocols. This procedure entails trimming raw reads derived from whole-genome or exome data followed by alignment to a known reference, post-alignment refinements, and variant calling. Trimmed reads are aligned to a reference by the Burrows-Wheeler Aligner (BWA), bwa-mem. Refinements of mismatches near insertions and deletions (indels) and base qualities are performed using GATK indels realignment and base recalibration to improve read quality after alignment. Processed reads are marked as fragment duplicates using Picard MarkDuplicates and single-nucleotide polymorphisms and small indels are identified using either GATK haplotype callers or SAMtools mpileup.
The Genome in a Bottle dataset was used to select steps and parameters minimizing the false-positive rate and maximizing the true-positive variants to achieve a sensitivity of 99.7%, precision of 99.1%, and F1 score of 99.4%. Finally, additional annotations are incorporated using dbNSFP and / or Gemini and QC metrics are collected at various stages and visualized using MultiQC.
Latest release 3.2.0 of GenPipes supports a new DNA sequencing protocol option called ‘sv’ besides the mugqic, mpileup and light options. This is useful for structural variation detection.
Structural Variation (SVs) are the genetic variations in the structure of chromosome with different types of rearrangements. They comprise millions of nucleotides of heterogeneity within every genome, and are likely to make an important contribution to genetic diversity and disease susceptibility. In the genomics community, substantial efforts have been devoted to improving understanding of the roles of SVs in genome functions relating to diseases and researchers are working actively to develop effective algorithms to reliably identify various types of SVs such as deletions, insertions, duplications and inversions. GenPipes supports SV detection option in the DNA Sequencing Pipeline.
Introduction¶
The standard MUGQIC DNA-Seq pipeline uses BWA to align reads to the reference genome. Treatment and filtering of mapped reads approaches as INDEL realignment, mark duplicate reads, recalibration and sort are executed using Picard and GATK. Samtools MPILEUP and bcftools are used to produce the standard SNP and indels variants file (VCF). Additional SVN annotations mostly applicable to human samples include mappability flags, dbSNP annotation and extra information about SVN by using published databases. The SNPeff tool is used to annotate variants using an integrated database of functional predictions from multiple algorithms (SIFT, Polyphen2, LRT and MutationTaster, PhyloP and GERP++, etc.) and to calculate the effects they produce on known genes. Refer to the list of SnpEff effects.
A summary html report is automatically generated by the pipeline. This report contains description of the sequencing experiment as well as a detailed presentation of the pipeline steps and results. Various Quality Control (QC) summary statistics are included in the report and additional QC analysis is accessible for download directly through the report. The report includes also the main references of the software and methods used during the analysis, together with the full list of parameters that have been passed to the pipeline main script.
The DNA sequencing pipeline supports two trimmers: Trimmomatic and Skewer.
GenPipes DNA sequencing pipeline offers four different protocol options:
The default protocol based on the GATK variant caller, haplotype caller, (-t mugqic)
Another based on the mpileup/bcftools caller (-t mpileup).
Light option (-t light)
Structural Variation Detection, sv option (-t sv)
See More information section below for details.
Version¶
3.6.2
For the latest implementation and usage details refer to DNA Sequencing implementation README file file.
Usage¶
dnaseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-t {mugqic,mpileup,light,sv}] [-r READSETS] [-v]
Optional Arguments
-t {mugqic,mpileup,light, sv}, --type {mugqic,mpileup,light,sv}
DNAseq analysis type
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute MUGQIC DNA sequencing pipeline:
dnaseq.py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/dnaseq/dnaseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/dnaseq/dnaseq.guillimin.ini -r readset.dnaseq.txt -s 1-29 -g dnaseqCommands_mugqic.sh
bash dnaseqCommands_mugqic.sh
Use the following commands to execute the Mpileup DNA sequencing pipeline:
dnaseq.py -t mpileup -c $MUGQIC_PIPELINES_HOME/pipelines/dnaseq/dnaseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/dnaseq/dnaseq.guillimin.ini -r readset.dnaseq.txt -s 1-33 -g dnaseqCommands_mpileup.sh
bash dnaseqCommands_mpileup.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline here.
Pipeline Schema¶
Figure below shows the schema of the DNA sequencing protocol - MUGQIC type.
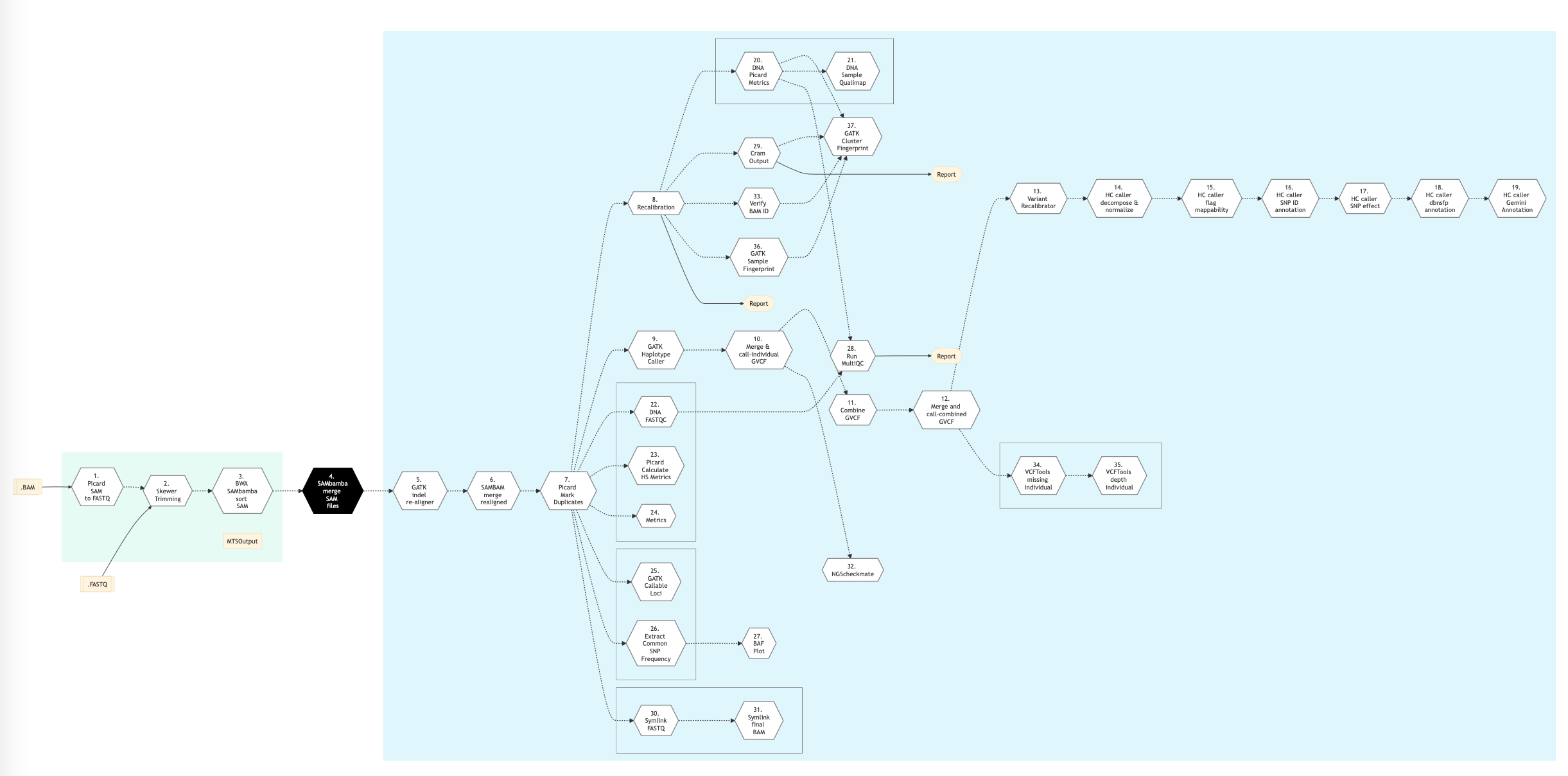
Figure: Schema of MUGQIC DNA Sequencing protocol¶
The following figure shows the pipeline schema for Mpileup type of DNA sequencing protocol.
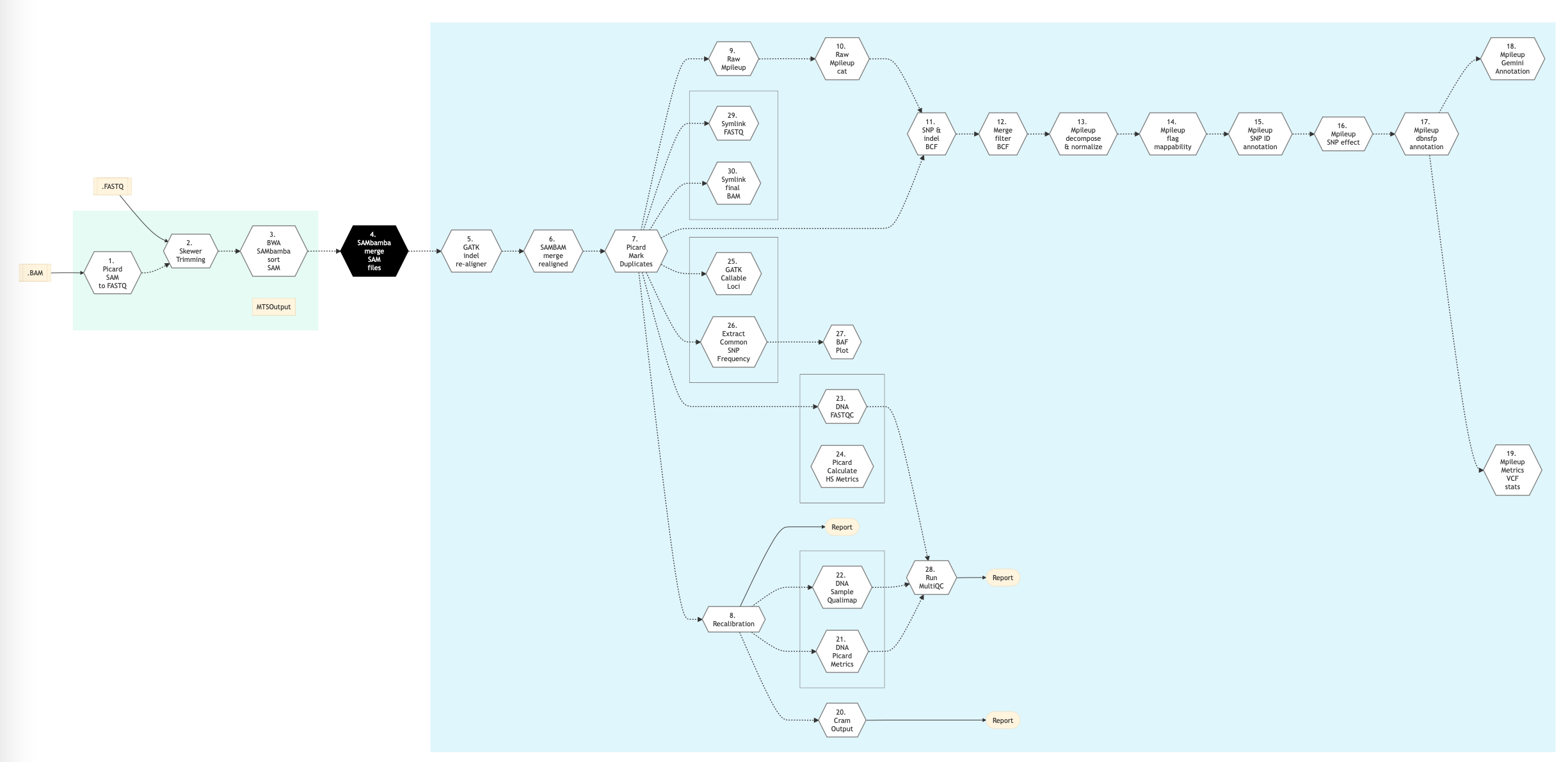
Figure: Schema of Mpileup DNA Sequencing protocol¶
The following figure shows the pipeline schema for ‘Light’ type of DNA sequencing protocol.
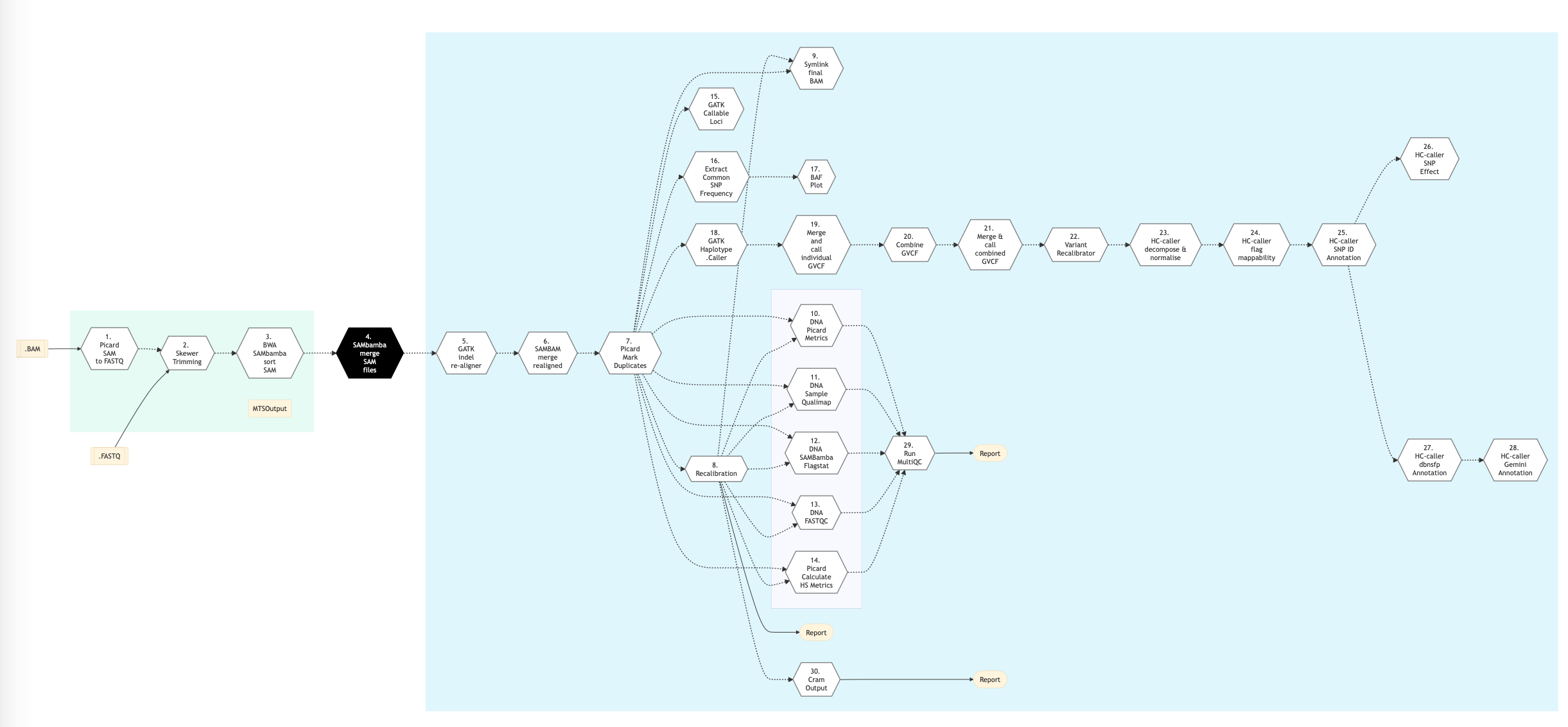
Figure: Schema of Light DNA Sequencing protocol¶
The following figure shows the pipeline schema for Structural Variation type of DNA sequencing protocol.
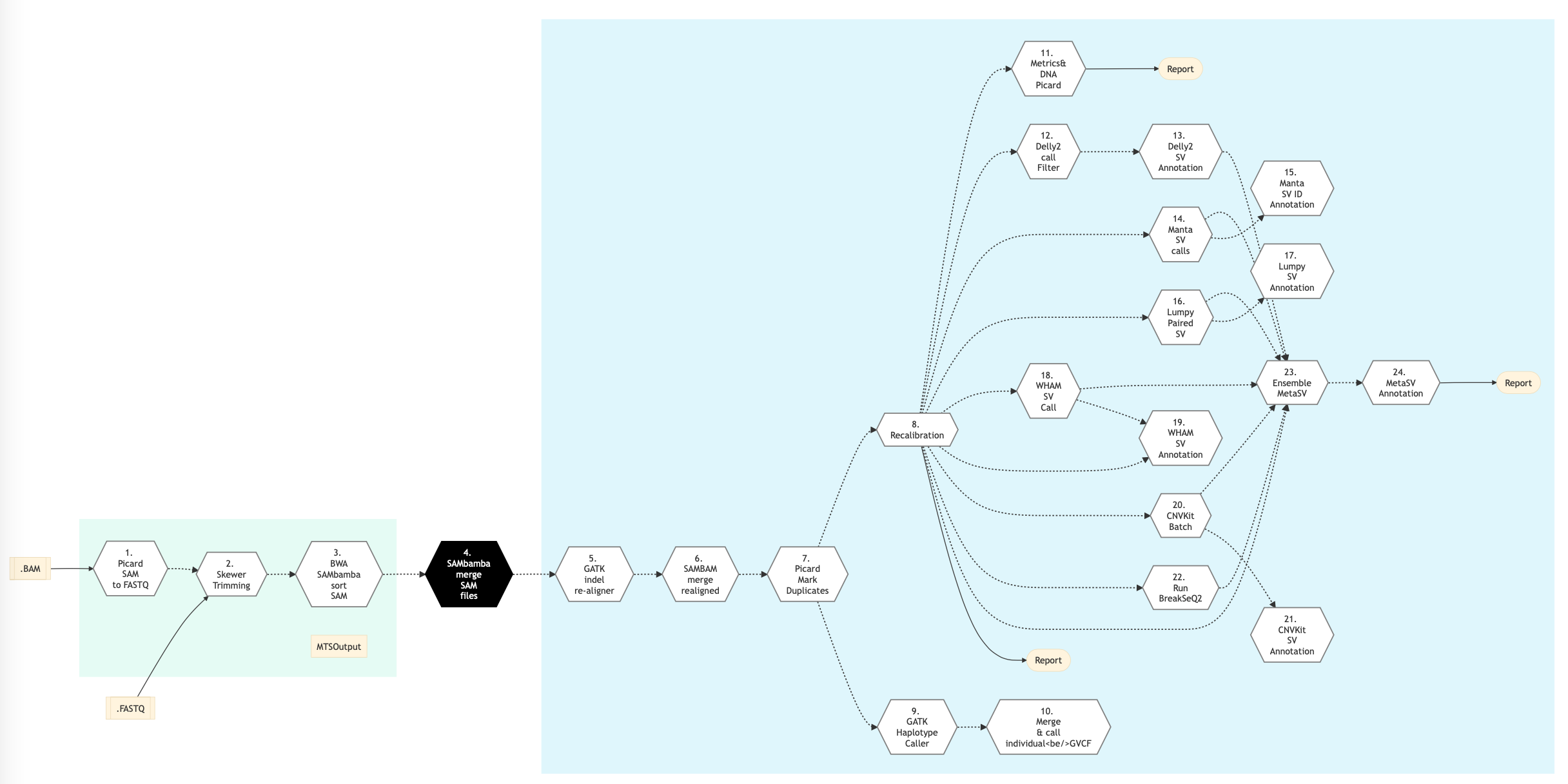
Figure: Schema of Structural Variations (SV) DNA Sequencing protocol¶
Refer to the Pipeline steps below, for details.
Pipeline Steps¶
The table below shows various steps that constitute the MUGQIC and Mpileup types of DNA Sequencing for genomic analysis pipelines.
MUGQIC DNA Sequencing Steps |
MPileup DNA Sequencing Steps |
Light DNA Sequencing Steps |
Structural Variations Steps |
|
|---|---|---|---|---|
30 |
||||
Step Details¶
Following are the various steps that are part of GenPipes DNA Sequencing genomic analysis pipeline:
Picard SAM to FastQ
Convert SAM/BAM files from the input readset file into FASTQ format if FASTQ files are not already specified in the readset file. Do nothing otherwise.
Sym Link FastQ
TBD-GenPipes-Dev
Trimmomatic
If available, this step takes FastQ file from the readset as input. Otherwise, FastQ output files from the previous Picard SAM to FastQ step, where BAM files are converted to FastQ format, are utilized in this step.
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Stats
The trim statistics per readset file are merged in this step.
GATK Indel Re-aligner
Insertion and deletion realignment is performed on regions where multiple base mismatches are preferred over indels by the aligner since it can appear to be less costly by the algorithm. Such regions will introduce false positive variant calls which may be filtered out by realigning those regions properly. Realignment is done using GATK Software. The reference genome is divided by a number regions given by the nb_jobs parameter.
Fix Mate by Coordinate
This step fixes the read mates. Once local regions are realigned, the read mate coordinates of the aligned reads need to be recalculated since the reads are realigned at positions that differ from their original alignment. Fixing the read mate positions is done using`BVATools`_.
Picard Mark Duplicates
This step marks duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be marked as a duplicate in the BAM file. Marking duplicates is done using Picard.
Recalibration
This step is used to recalibrate base quality scores of sequencing-by-synthesis reads in an aligned BAM file. After recalibration, the quality scores in the QUAL field in each read in the output BAM are more accurate in that the reported quality score is closer to its actual probability of mismatching the reference genome. Moreover, the recalibration tool attempts to correct for variation in quality with machine cycle and sequence context, and by doing so, provides not only more accurate quality scores but also more widely dispersed ones.
Sym Link Final BAM
TBD-GenPipes-Dev
Metrics DNA Picard
TBD-GenPipes-Dev
Metrics DNA Sample Quality Map
TBD-GenPipes-Dev
Metrics DNA SAMBAM Flag Stats
TBD-GenPipes-Dev
Metrics DNA FastQC
TBD-GenPipes-Dev
Picard Calculate HS Metrics
Compute on target percent of hybridization based capture happens in this step.
GATK Callable Loci
Computes the callable region or the genome as a bed track.
Extract Common SNP Frequencies
Extracts allele frequencies of possible variants across the genome.
BAF Plot
Plots DepthRatio and B allele frequency of previously extracted alleles.
GATK Haplotype Caller
GATK Haplotype Caller is used for SNPs and small indels.
Merge and call individual GVCF
Merges the gvcfs of haplotype caller and also generates a per sample vcf containing genotypes.
Combine GVCF
Combine the per sample gvcfs of haplotype caller into one main file for all sample.
Merge and call combined GVCF
Merges the combined gvcfs and also generates a general vcf containing genotypes.
Variant Recalibrator
This step involves GATK variant recalibrator. The purpose of the variant recalibrator is to assign a well-calibrated probability to each variant call in a call set. You can then create highly accurate call sets by filtering based on this single estimate for the accuracy of each call. The approach taken by variant quality score recalibration is to develop a continuous, covarying estimate of the relationship between SNP call annotations (QD, MQ, HaplotypeScore, and ReadPosRankSum, for example) and the probability that a SNP is a true genetic variant versus a sequencing or data processing artifact. This model is determined adaptively based on “true sites” provided as input, typically HapMap 3 sites and those sites found to be polymorphic on the Omni 2.5M SNP chip array. This adaptive error model can then be applied to both known and novel variation discovered in the call set of interest to evaluate the probability that each call is real. The score that gets added to the INFO field of each variant is called the VQSLOD. It is the log odds ratio of being a true variant versus being false under the trained Gaussian mixture model. Using the tranche file generated by the previous step the ApplyRecalibration walker looks at each variant’s VQSLOD value and decides which tranche it falls in. Variants in tranches that fall below the specified truth sensitivity filter level have their filter field annotated with its tranche level. This will result in a call set that simultaneously is filtered to the desired level but also has the information necessary to pull out more variants for a higher sensitivity but a slightly lower quality level.
Haplotype caller decompose and normalize
TBD-GenPipes-Dev
Haplotype caller flag mappability
Mappability annotation applied to haplotype caller vcf. An in-house database identifies regions in which reads are confidently mapped to the reference genome.
Haplotype caller SNP ID annotation
dbSNP annotation applied to haplotype caller vcf. The .vcf files are annotated for dbSNP using the software SnpSift (from the SNPEff Suite).
Haplotype caller SNP Effect
Variant effect annotation applied to haplotype caller vcf. The .vcf files are annotated for variant effects using the SNPEff Suite software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes).
Haplotype caller dbNSFP annotation
Additional SVN annotations applied to haplotype caller vcf. Provides extra information about SVN by using numerous published databases. Applicable to human samples. Databases available include Biomart (adds GO annotations based on gene information) and dbNSFP (an integrated database of functional annotations from multiple sources for the comprehensive collection of human non-synonymous SNPs. It compiles prediction scores from four prediction algorithms (SIFT, Polyphen2, LRT and MutationTaster), three conservation scores (PhyloP, GERP++ and SiPhy) and other function annotations).
Haplotype caller Gemini annotation
TBD-GenPipes-Dev
Haplotype caller metrics VCF stats
Metrics SNV applied to haplotype caller vcf. Multiple metrics associated to annotations and effect prediction are generated at this step: change rate by chromosome, changes by type, effects by impact, effects by functional class, counts by effect, counts by genomic region, SNV quality, coverage, InDel lengths, base changes, transition-transversion rates, summary of allele frequencies, codon changes, amino acid changes, changes per chromosome, change rates.
Run MultiQC
TBD-GenPipes-Dev
Raw MPileup
Full pileup (optional). A raw mpileup file is created using samtools mpileup and compressed in gz format. One packaged mpileup file is created per sample/chromosome.
Compress Raw MPileup
Merge mpileup files per sample/chromosome into one compressed gzip file per sample.
SNP and indel BCF
Mpileup and Variant calling. Variants (SNPs and INDELs) are called using SAMTools software package mpileup. bcftools view is used to produce binary bcf files.
Merge Filter BCF
bcftools is used to merge the raw binary variants files created in the snpAndIndelBCF step. The output of bcftools is fed to varfilter, which does an additional filtering of the variants and transforms the output into the VCF (.vcf) format. One vcf file contain the SNP/INDEL calls for all samples in the experiment.
MPileup decompose and normalize
TBD-GenPipes-Dev
MPileup Flag Mappability
Mappability annotation applied to mpileup vcf. An in-house database identifies regions in which reads are confidently mapped to the reference genome.
Mpileup SNP ID annotation
dbSNP annotation applied to MPileup vcf. The .vcf files are annotated for dbSNP using the software SnpSift (from the SNPEff Suite).
MPileup SNP Effect
Variant effect annotation applied to mpileup vcf. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes).
MPileup dbNSFP annotation
Additional SVN annotations applied to mpileup vcf. Provides extra information about SVN by using numerous published databases. Applicable to human samples. Databases available include Biomart (adds GO annotations based on gene information) and dbNSFP (an integrated database of functional annotations from multiple sources for the comprehensive collection of human non-synonymous SNPs. It compiles prediction scores from four prediction algorithms (SIFT, Polyphen2, LRT and MutationTaster), three conservation scores (PhyloP, GERP++ and SiPhy) and other function annotations).
MPileup Gemini annotation
TBD-GenPipes-Dev
MPileup Metrics VCF stats
Metrics SNV applied to mpileup caller vcf. Multiple metrics associated to annotations and effect prediction are generated at this step: change rate by chromosome, changes by type, effects by impact, effects by functional class, counts by effect, counts by genomic region, SNV quality, coverage, InDel lengths, base changes, transition-transversion rates, summary of allele frequencies, codon changes, amino acid changes, changes per chromosome, change rates.
SAMBAM Mark Duplicates
In this step duplicates are marked. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in case of paired-end reads). All but the best pair (based on alignment score) will be marked as duplicates in the BAM file. Marking duplicates is done using Picard.
Delly2 Call Filter
This step uses normal and tumor final BAMs as input and generates a binary variant call format (BCF) file as output. It utilizes Delly2, an integrated structural variant (SV) prediction method that can discover genotype and visualize deletions, tandem duplications, inversions and translocations at single-nucleotide resolution in short-read massively parallel sequencing data. It uses paired-ends, split-reads and read-depth to sensitively and accurately delineate genomic rearrangements throughout the genome. Structural variants can be annotated using Delly-sansa and visualized using Delly-maze or Delly-suave.
Delly2 SV Annotation
SV Annotation step utilizes the BCF file generated in previous Delly2 Call Filter step and performs genome annotation at various levels. At the nucleotid level it tries to identify the physical location of the SV dna sequences. Next, at the protein level the annotation process tries to determine the possible functions of the SV genes. Lastly, at the process-level annotation, it tries to identify the pathways and processes in which different SV genes interact, assembling an efficient functional annotation. For more details on annotation see Genome Annotations.
Manta SV Calls
Manta calls structural variants (SVs) and indels from mapped paired-end sequencing reads. It is optimized for analysis of germline variation in small sets of individuals and somatic variation in tumor/normal sample pairs. Manta discovers, assembles and scores large-scale SVs, medium-sized indels and large insertions within a single efficient workflow.
Manta accepts input read mappings from BAM or CRAM files and reports all SV and indel inferences in VCF 4.1 format
** Manta SV Annotation**
This step uses the VCF file generated in previous step and performs SV annotations to compares types and breakpoints for candidate SVs from different callsets and enables fast comparison of SVs to genomic features such as genes and repetitive regions, as well as to previously established SV datasets.
Lumpy Paired SV
This step uses Lumpy for structural variant discovery in the given input file. The output is available in BAM format.
Comprehensive discovery of structural variation (SV) from whole genome sequencing data requires multiple detection signals including read-pair, split-read, read-depth and prior knowledge. Owing to technical challenges, extant SV discovery algorithms either use one signal in isolation, or at best use two sequentially. Lumpy is a novel SV discovery framework that naturally integrates multiple SV signals jointly across multiple samples. It yields improved sensitivity, especially when SV signal is reduced owing to either low coverage data or low intra-sample variant allele frequency.
Lumpy SV Annotation
This step performs LumPy SV Annotation for mapping and characterization of SVs.
Wham SV Call
Wham (Whole-genome Alignment Metrics) provides a single, integrated framework for both structural variant calling and association testing, thereby bypassing many of the difficulties that currently frustrate attempts to employ SVs in association testing. This step returns a VCF file.
Wham SV Annotation
This step uses the VCF file generated in the previous step Wham SV Call and performs SV annotations.
CNVkit Batch
A copy number variation (CNV) is when the number of copies of a particular gene varies from one individual to the next. Copy-number variation (CNV) is a large category of structural variation, which includes insertions, deletions and duplications. For copy number variation analysis, GenPipes DNA Sequencing pipeline (-t sv option) uses CNVkit that allows for CNV calling on single samples (e.g., tumor samples).
CNVkit provides an advantageous way to run the entire pipeline using the batch option to run various stages in copy number calling pipeline such as:
Create target/anti-target bed files
Gather read depths for those regions
Compile a copy number reference
Correct biases in tumor samples while calculating copy ratios
Mark copy number segments
CNVkit SV Annotation
This step performs CNVkit SV annotation.
Run BreakSeq2
In this step, BreakSeq2 is used to combine DNA double-strand breaks (DSBs) labeling with next generation sequencing (NGS) to map chromosome breaks with improved sensitivity and resolution. It is an ultra fast and accurate nucleotide-resolution analysis of structural variants.
Ensemble MetaSV
MetaSV uses highly effective ensemble approach for calling SVs. It is an integrated SV caller which leverages multiple orthogonal SV signals for high accuracy and resolution. MetaSV proceeds by merging SVs from multiple tools for all types of SVs. It also analyzes soft-clipped reads from alignment to detect insertions accurately since existing tools underestimate insertion SVs. Local assembly in combination with dynamic programming is used to improve breakpoint resolution. Paired-end and coverage information is used to predict SV genotypes.
MetaSV Annotation
This step uses output from previous step and performs SV annotations.
Metrics
This step computes metrics and generates coverage tracks per sample. Multiple metrics are computed at this stage:
Number of raw reads,
Number of filtered reads,
Number of aligned reads,
Number of duplicate reads,
Median, mean and standard deviation of insert sizes of reads after alignment,
Percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads)
Whole genome or targeted percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads).
A TDF (.tdf) coverage track is also generated at this step for easy visualization of coverage in the IGV browser.
Metrics NGSCheckmate
NGSCheckMate is a software package for identifying next generation sequencing (NGS) data files from the same individual. It analyzes various types of NGS data files including (but not limited to) whole genome sequencing (WGS), whole exome sequencing (WES), RNA-seq, ChIP-seq, and targeted sequencing of various depths. Data types can be mixed (e.g. WES and RNA-seq, or RNA-seq and ChIP-seq). It takes BAM (reads aligned to the genome), VCF (variants) or FASTQ (unaligned reads) files as input. NGSCheckMate uses depth-dependent correlation models of allele fractions of known single-nucleotide polymorphisms (SNPs) to identify samples from the same individual.
This step takes as input the file containing all vcfs in project output.
Metrics Verify BAM ID
In this step, VerifyBAMID software is used to verify whether the reads in particular file match previously known genotypes for an individual (or group of individuals), and checks whether the reads are contaminated as a mixture of two samples.
Metrics VCFTools Missing Individual
This step uses VCFtools and –missing_indv option to generate a file reporting the missingness factor in the analysis on a per-individual basis. It takes bgzipped .vcf file as input and creates .imiss flat file indicating missingness.
Metrics VCFTools Depth Individual
This step uses VCFtools and –depth option to generate a file containing the mean depth per individual. It takes as input bgzipped .vcf file and creates a .idepth flat file.
Metrics GATK Sample Fingerprint
CrosscheckFingerprints (Picard) functionality in GATK toolkit is used to cross-check readgroups, libraries, samples, or files to determine if all data in the set of input files appears to come from the same individual. In this step, sample SAM/BAM or VCF file is taken as input and a fingerprint file is generated using CrosscheckFingerprints (Picard) in GATK. It checks the sample identity of the sequence/genotype data in the provided file (SAM/BAM or VCF) against a set of known genotypes in the supplied genotype file (in VCF format).
Metrics GATK Cluster Fingerprint
In this step, ClusterCrosscheckMetrics function from GATK is used as a follow-up step to running CrosscheckFingerprints created in the Metrics GATK Sample Fingerprint step earlier. There are cases where one would like to identify a few groups out of a collection of many possible groups (say to link a bam to it’s correct sample in a multi-sample vcf. In this step, sample SAM/BAM or VCF file is taken as input and a fingerprint file is generated.
Skewer Trimming
TBD-GenPipes-Dev
BWA SAMbamba Sort SAM
The input for this step is the trimmed FASTQ files if available. Otherwise, it uses the FASTQ files from the readset. If those are not available then it uses FASTQ output files from the previous ‘Picard SAM to FASTQ`_ step where BAM files are converted to FASTQ format.
In this step, filtered reads are aligned to a reference genome. The alignment is done per sequencing readset. The alignment software used is BWA with algorithm: bwa mem. BWA output BAM files are then sorted by coordinate using SAMBAMBA.
SAMBAM Merge SAM Files
This step takes as input files:
Aligned and sorted BAM output files from previous bwa_mem_picard_sort_sam step if available
Else, BAM files from the readset file
In this step, BAM readset files are merged into one file per sample. Merge is done using Picard.
SAMBAM Merge Realigned
In this step, BAM files of regions of realigned reads are merged per sample using SAMBAMBA.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to DNA Sequencing implementation README.md file.
dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions
DNA-Seq Pipeline: some interesting information.
Manta - Rapid Detection of Structural Variants, Using Manta for filtering and annotations.