CoV Sequencing Pipeline¶
CoV Sequencing Pipeline can be used to effectively amplify and detect SARS-CoV-2 RNA in samples such that it enables reliable results from even low copy numbers. It helps to assay clean characteristic target peaks of defined sizes, allowing for direct detection of the presence of viral genome from the Coronaviridae family. Sequencing provides confirmation for the species as well as phylogenetic information for the specific strain discrimination.
The design of this pipeline is directed against SARS-CoV-2 and other coronaviruses as well. By amplifying conserved regions of other coronaviruses in a sample, along with mutation tolerant panels, it can provide additional insights and pinpoint sequence variability, thus offering a powerful solution for more in-depth research and surveillance of the rapidly evolving virus.
CoVSeQ pipeline is designed as part of the partnership for Québec SARS-CoV-2 sequencing. It is funded by the CanCOGeN initiative through Genome Canada and from the Ministere de la santé et des services sociaux du Québec. For more details, see CoVSeQ website.
Introduction¶
TBD some high level introduction about Pipeline steps et all and how it is different from other available CoV Seq pipelines (if any). Refer to Ed and Hector.
Version¶
3.6.2
For the latest implementation and usage details refer to DNA Sequencing implementation README file file.
Usage¶
covseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>]
[--genpipes_file GENPIPES_FILE]
[-r READSETS] [-v]
Optional Arguments
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute CoVSeq sequencing pipeline:
covseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/covseq/covseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/covseq/covseq.beluga.ini -g covseqCommands_mugqic.sh
bash covseqCommands_mugqic.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline here.
Note
Check with Paul/Hector if this pipeline requires a test dataset and whether one is available. Then update/edit the test dataset link above accordingly.
Pipeline Schema¶
Figure below shows the schema of the CovSeq sequencing protocol.
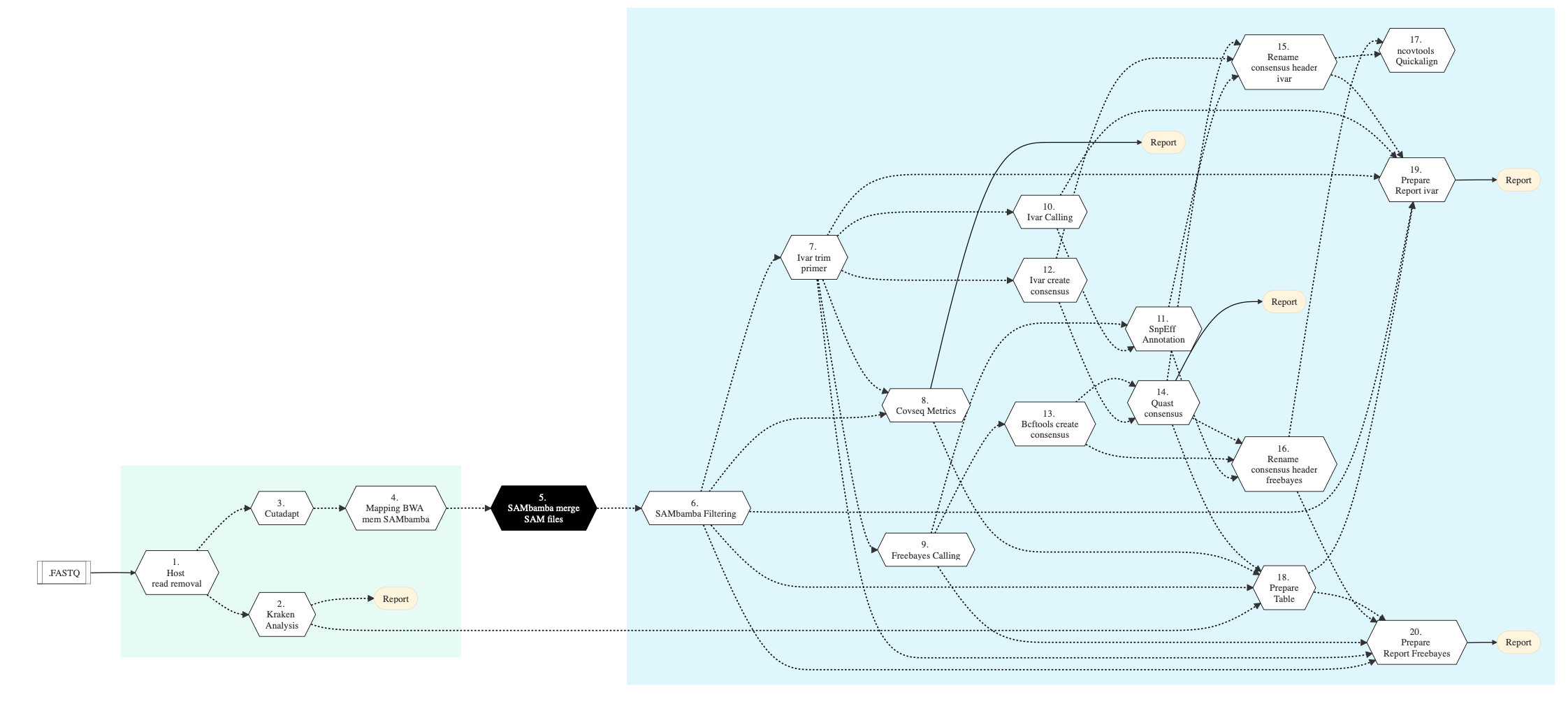
Figure: Schema of CoVSeq Sequencing protocol¶
Pipeline Steps¶
The table below shows various steps that constitute the CoVSeq genomic analysis pipeline.
SARS-CoV-2 Sequencing Steps |
|
|---|---|
Step Details¶
Following are the various steps that are part of GenPipes CoVSeq genomic analysis pipeline:
Host Reads Removal
In this step, the filtered reads are aligned to a reference genome. The alignment is done per sequencing readset using the BWA Software and BWA-Mem Algorithm. BWA output BAM files are then sorted by coordinate using Sambamba.
Input files for this step include:
Trimmed FASTQ files, if available
If no FASTQ files then FASTQ files from supplied readset is used.
If no readset is supplied then FASTQ output from Picard SAM to FASTQ conversion of BAM files is used.
Kraken Analysis
Kraken is an ultra fast and highly accurate mechanism for assigning taxonomic labels to metagenomic DNA sequences. It achieves high sensitivity and speed by utilizing exact alignments of k-mers and a novel classification algorithm. This step performs Kraken analysis using output of the Sambamba processing in previous step.
Cutadapt
Cutadapt processing cleans data by finding and removing adapter sequences, primers, poly-A tails and other types of unwanted sequence from high throughput sequencing reads obtained after Kraken analysis. In this step, quality trimming of raw reads and removing of adapters is performed by giving ‘Adapter1’ and ‘Adapter2’ columns from the readset file to Cutadapt. For PAIRED_END readsets, both adapters are used. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
To trim the front of the read, use adapter_5p_fwd and adapter_5p_rev (For PAIRED_END only) in cutadapt section of the .ini file.
This step takes as input files:
FASTQ files from the readset file, if available.
Otherwise, FASTQ output files from previous Picard SAM to FASQ conversion of BAM files is used.
Mapping BWA Mem Sambamba
This step takes as input files trimmed FASTQ files, if available. Otherwise it takes FASTQ files from the readset. If readset is not supplied then it uses FASTQ output files from the previous Picard SAM to FASTQ conversion of BAM files.
Here, the filtered reads are aligned to a reference genome. The alignment is done per sequencing readset. BWA Software is used for alignment with BWA-Mem Algorithm. BWA output BAM files are then sorted by coordinate using Sambamba.
Sambamba Merge SAM Files
This step uses Sambamba-Merge Tool to merge several BAM files into one. SAM headers are merged automatically similar to how it is done in Picard merging tool.
Sambamba Filtering
In this step, raw BAM files are filtered using Sambamba and and `awk` command is run to filter by insert size.
iVar Trim Primers
iVar uses primer positions supplied in a BED file to soft clip primer sequences from an aligned and sorted BAM file. Following this, the reads are trimmed based on a quality threshold(Default: 20). To do the quality trimming, iVar uses a sliding window approach(Default: 4). The windows slides from the 5’ end to the 3’ end and if at any point the average base quality in the window falls below the threshold, the remaining read is soft clipped.
In this step, primer sequences are removed from individual BAM files using iVar.
CoVSeq Metrics
In this step, multiple metrics are computed from sequencing:
DNA Sample Qualimap to facilitate quality control of alignment sequencing
Sambamba-flagstat for obtaining flag statistics from BAM file
BED Tools GenomeCov is used for computing histograms(default), per-base reports and BEDGRAPH summaries of feature coverage of aligned sequences for a given genome.
Picard HS Metrics are picked from SAM/BAM files. Only those metrics are collected that are specific for sequence datasets generated through hybrid-selection. Hybrid-selection (HS) is the most commonly used technique to capture exon-specific sequences for targeted sequencing experiments such as exome sequencing.
Freebayes Calling
Freebayes is a haplotype-based variant detector designed to find small polymorphisms, specifically SNPs (single-nucleotide polymorphisms), indels (insertions and deletions), MNPs (multi-nucleotide polymorphisms), and complex events (composite insertion and substitution events) smaller than the length of a short-read sequencing alignment.
This method avoids one of the core problems with alignment-based variant detection— that identical sequences may have multiple possible alignments. See Freebayes details here.
iVar Calling
In this step, iVar is used for creating a trimmed BAM file after trimming aligned reads in the input BAM file using primer positions specified in the BED input file. Besides the Freebayes Calling tool, ivar calling is also used in covseq pipeline as part of the latest release.
SNPEff Annotate
This step uses SNPEff to annotate the data.
iVar Create Consensus
In this step, iVar is used to create consensus. Also, it removes primer sequences to individual BAM files using fgbio.
BCFTools Create Consensus
BCFtools consensus is created in this step. BCFtools is a set of utilities that manipulate variant calls in the Variant Call Format (VCF) and its binary counterpart BCF
QUAST Consensus Metrics
In this step, QUAST is used to compare and evaluate assemblies to rule out misassemblies.
Rename Consensus Header ivar
Consensus sequence is the calculated order of most frequent residues found at each position in a sequence alignment. This information is important when considering sequence-dependent enzymes such as RNA Polymerase which is important for SAR-CoV-2 studies. In this step, header sequence can be modified in various ways as specified in rename type parameter: Multipart header, Replace word, Replace interval, and Add prefix/suffix.
Rename Consensus Header freebayes
Two variant calling tools are used in covseq pipeline - ivar and freebayes. In this step, the consensus header rename for freebayes is done.
ncovtools Quickalign
Uses quickalign to provides summary statistics, which can be used to determine the sequencing quality and evolutionary novelty of input genomes (e.g. number of new mutations and indels).
It uses ivar consensus as well as freebayes consensus to arrive at the alignment decisions.
Prepare Table
Gathers all analysis data for quast, kraken and other metrics and module details.
Prepare Report ivar
Prepare ivar analysis report.
Prepare Report Freebayes
Prepare Freebayes analysis report.
More information¶
For the latest implementation and usage details refer to CoVSeq Pipeline implementation README.md.