Tumor Pair Sequencing Pipeline¶
Warning
A new updated Tumor Pair is back in the release 3.6.0
Please note that Tumor Pair Sequencing Pipeline was not available in GenPipes Release 3.2.0 until 3.6.0.
The documentation below refers to the pipeline corresponding to GenPipes Release 3.1.5.
We are in the process of updating the documentation corresponding to latest release 3.6.0. Thank you for your patience.
Human genome comprises of a set of chromosome pairs. One chromosome in each pair, called homolog, is derived from each parent. It is typically referred to as diploid whereas the set of chromosomes from a single parent is called haploid genome. For a given gene on a given chromosome, there is a comparable, if not identical, gene on the other chromosome in the pair, known as an allele. Large structural alterations in chromosomes can change the number of copies of affected genes on those chromosomes. This is one of the key reasons for causing cancer. In cancer cells, instead of having a homologous pair of alleles for a given gene, there may be deletions or duplications of those genes.
Such alterations leads to unequal contribution of one allele over the other, altering the copy number of a given allele. These variations in copy number indicated by the ratio of cancer cell copy number to normal cell copy number can provide information regarding the structure and history of cancer. However, when DNA is extracted, there is a mix of cancer and normal cells and this information regarding absolute copy number per cancer cell is lost in DNA extraction process. Hence it must be inferred.
Inferring absolute copy number is difficult for three reasons:
cancer cells are nearly always intermixed with an unknown fraction of normal cells; the measure for this is tumor purity
the actual quantity of DNA in the cancer cells after gross numerical and structural chromosomal changes is unknown; the measure for this is tumor ploidy
the cancer cell population may be heterogeneous, possibly because of ongoing mutations and changes
Tumor purity and ploidy have a substantial impact on next-gen sequence analyses of tumor samples and may alter the biological and clinical interpretation of results.
GenPipes Tumor Analysis pipeline is designed to detect somatic variants from a tumor and match normal sample pair more accurately.
Introduction¶
GenPipes Tumor Pair workflow consumes BAM files. It inherits the BAM processing protocol from DNA-seq implementation, for retaining the benchmarking optimizations, but differs in alignment refinement and mutation identification. It achieves this by maximizing the information, utilizing both tumor and normal samples together.
The pipeline is based on an ensemble approach, which was optimized using both the DREAM3 challenge and the CEPH mixture datasets to select the best combination of callers for both SNV and structural variation detection. For SNVs, multiple callers such as ‘GATK MuTect2`_, VarScan 2, BCFTools, VarDict were combined to achieve a sensitivity of 97.5%, precision of 98.8%, and F1 score of 98.1% for variants found in ≥2 callers.
Similarly, SVs were identified using multiple callers such as DELLY, LUMPY, WHAM, CNVKit, and SvABA combined using MetaSV to achieve a sensitivity of 84.6%, precision of 92.4%, and F1 score of 88.3% for duplication variants found in the DREAM3 dataset. The pipeline also integrates specific cancer tools to estimate tumor purity and tumor ploidy of sample pair normal−tumor.
Additional annotations are incorporated to the SNV calls using dbNSFP and/or Gemini, and QC metrics are collected at various stages and visualized using MultiQC.
GenPipes Tumor Pair pipeline has three protocol options: sv, ensemble, or fastpass. For details refer to Pipeline Schema section below.
Version¶
3.6.2
For the latest implementation and usage details refer to Tumor Pair Pipeline implementation README file file.
Usage¶
python tumor_pair.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-p PAIRS] [-t {sv,ensemble,fastpass}] [-r READSETS] [-v]
Optional Arguments
-p PAIRS, --pairs PAIRS
pairs file
-t {sv,ensemble,fastpass}, --type {sv,ensemble,fastpass}
tumor pair analysis type
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute MUGQIC DNA sequencing pipeline:
python tumor_pair.py -c $MUGQIC_PIPELINES_HOME/pipelines/dnaseq/dnaseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/tumor_pair/tumor_pair.base.ini $MUGQIC_PIPELINES_HOME/pipelines/tumor_pair/tumor_pair.guillimin.ini -r readset.tumorPair.txt -p pairs.csv -s 1-44 -g tumor_pairCommands.sh
bash tumor_pairCommands.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
where, p pairs : format - patient_name,normal_sample_name,tumor_sample_name
You can download the test dataset for this pipeline here.
Pipeline Schema¶
Pair Pipeline. You can refer to the latest pipeline implementation.
There are three options for Tumor Pair Pipeline: sv, ensemble and fastpass.
Figure below shows the schema of the Tumor Pair Pipeline (sv) option. See here to download a high resolution image of Tumor Pair Sequencing Pipeline (sv) to download a high resolution image of the same.
{kind=link}
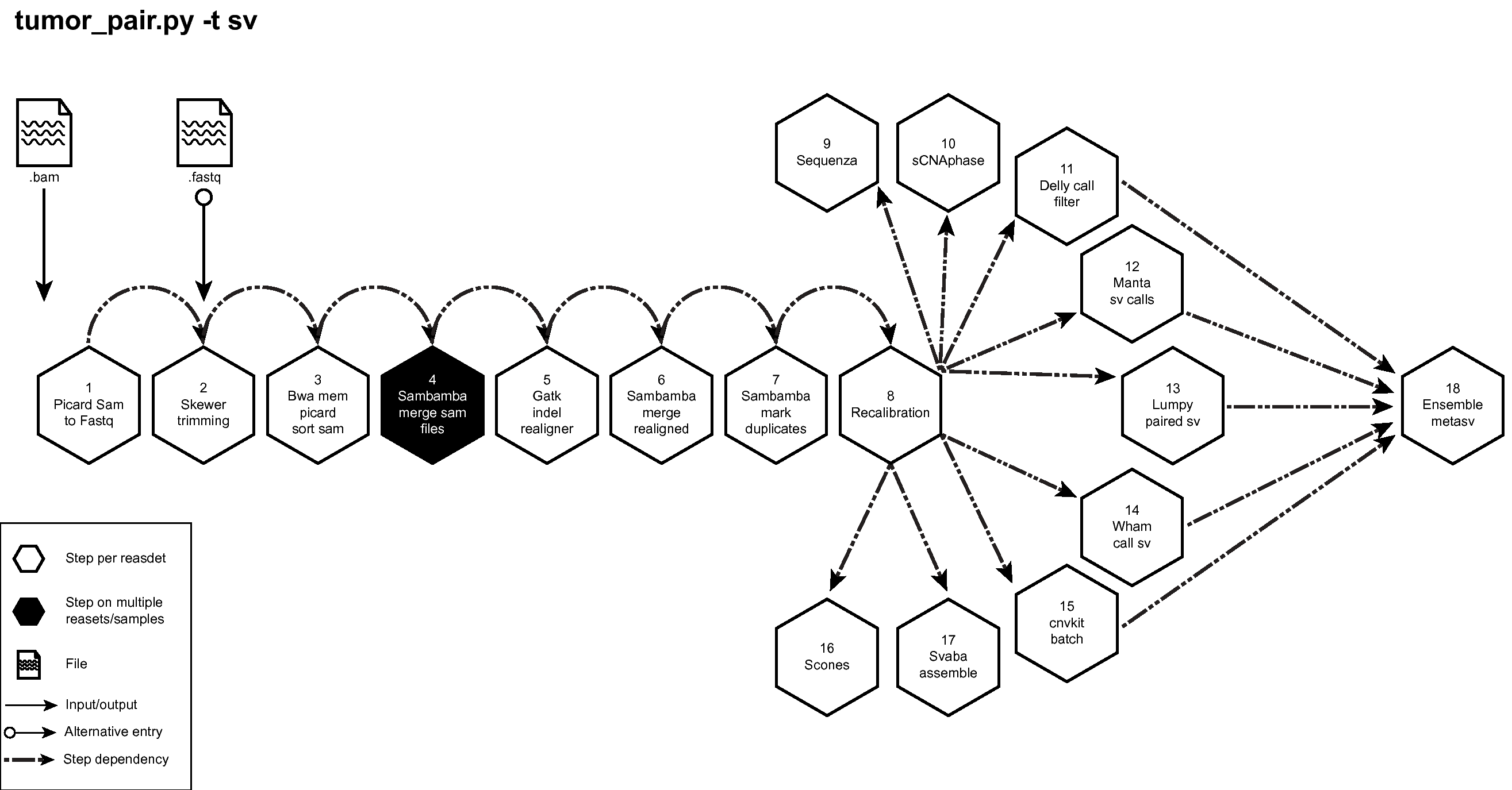
Figure: Schema of Tumor Pair Pipeline (sv)¶
Figure below shows the schema of the Tumor Pair Pipeline (ensemble) option. See here to download a high resolution image of Tumor Pair Sequencing Pipeline (ensemble) to download a high resolution image of the same.
{kind=link}
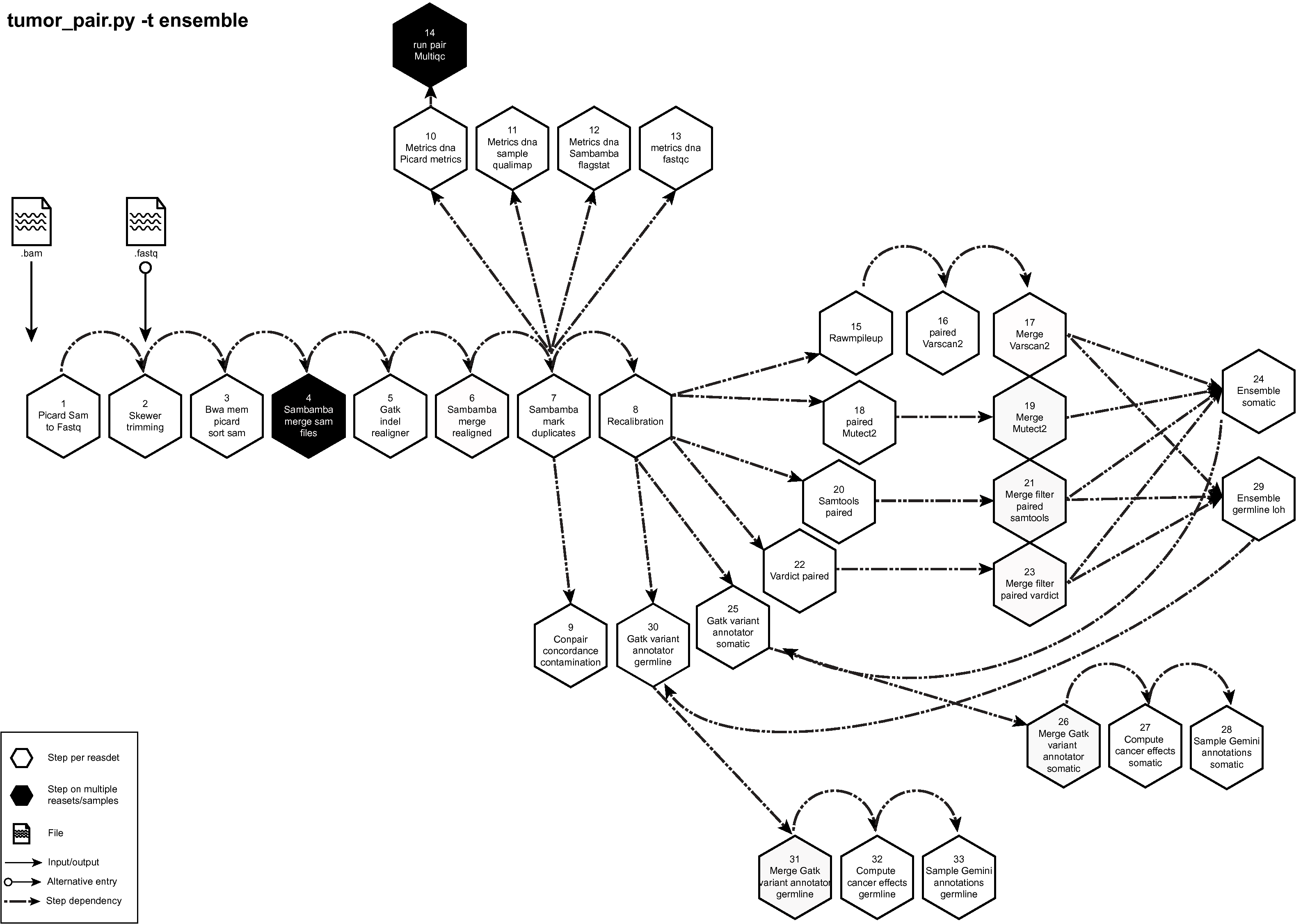
Figure: Schema of Tumor Pair Pipeline (ensemble)¶
Figure below shows the schema of the Tumor Pair Pipeline (fastpass) option. See here to download a high resolution image of Tumor Pair Sequencing Pipeline (fastpass) to download a high resolution image of the same.
{kind=link}
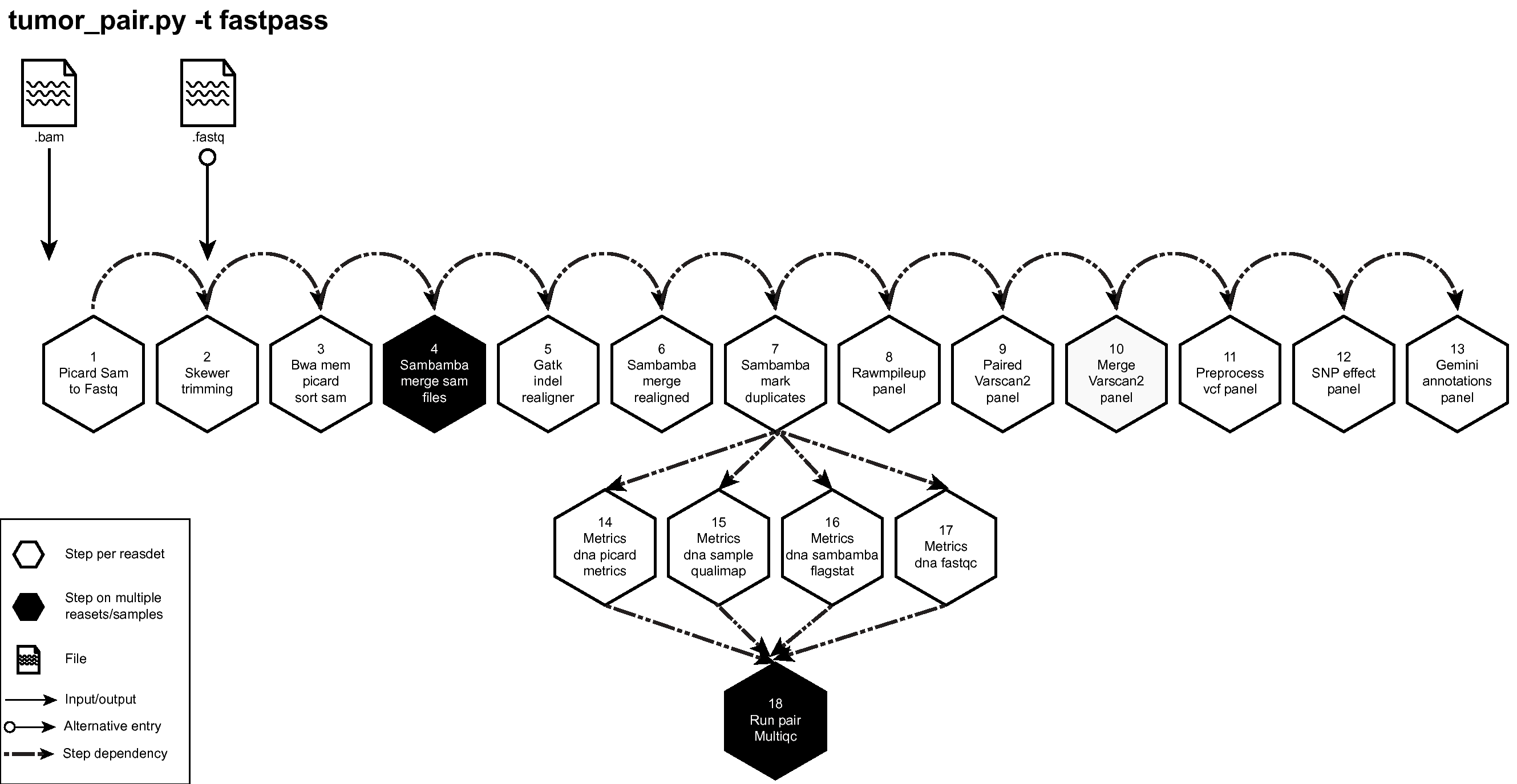
Figure: Schema of Tumor Pair Pipeline (fastpass)¶
Pipeline Steps¶
The table below shows various steps that constitute the Tumor Pair Pipeline.
Tumor Pair Pipeline Steps |
|
|---|---|
Step Details¶
Following are the various steps that are part of GenPipes Tumor Pair Pipeline:
Picard SAM to FastQ
This step converts SAM/BAM files from the input readset file into FASTQ format, if FASTQ files are not already specified in the readset file. Otherwise, it does nothing.
Trimmomatic
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic Method. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Stats
The trim statistics per readset are merged at this step.
BWA Picard Sort SAM
This step takes as input files:
Trimmed FASTQ files if available
Else, FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
The filtered reads are aligned to a reference genome. The alignment is done per sequencing readset. The alignment software used is BWA with algorithm: bwa mem. BWA output BAM files are then sorted by coordinate using Picard.
SamBamba Merge Files
This step takes as input files:
Aligned and sorted BAM output files from previous BWA Picard Sort step, if available
Else, BAM files from the readset file
BAM readset files are merged into one file per sample. Merge is done using Picard.
GATK InDel Realigner
Insertion and deletion realignment is performed on regions where multiple base mismatches are preferred over indels by the aligner since it can appear to be less costly by the algorithm. Such regions will introduce false positive variant calls which may be filtered out by realigning those regions properly. Realignment is done using GATK. The reference genome is divided by a number regions given by the nb_jobs parameter.
Note: modified to use both normal and tumor BAMs to reduce FPs around indels
SamBamba Merge Realigned
BAM files of regions of realigned reads are merged per sample using SamBamba.
Sambamba Mark Duplicates
Mark duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be marked as a duplicate in the BAM file. Marking duplicates is done using Picard.
Recalibration
This step re-calibrates base quality scores of sequencing-by-synthesis reads in an aligned BAM file. After recalibration, the quality scores in the QUAL field in each read in the output BAM are more accurate in that the reported quality score is closer to its actual probability of mismatching the reference genome. Moreover, the recalibration tool attempts to correct for variation in quality with machine cycle and sequence context, and by doing so, provides not only more accurate quality scores but also more widely dispersed ones.
Conpair Concorance Contamination
Conpair is a concordance and contamination estimator for tumor–normal pairs. This step is a quality control process to ensure the normal sample and cancer sample come from the same patient. It estimates this by determining the amount of normal sample in the tumor and the amount of tumor in normal sample.
Raw Mpileup Panel
Full pileup (optional). A raw Mpileup file is created using samtools Mpileup and compressed in gzipped format. One packaged Mpileup file is created per sample/chromosome.
Paired VarScan 2
This step involves variant calling and somatic mutation/CNV detection for next-generation sequencing data. For details see - VarScan 2 Somatic mutation and copy number alteration discovery in cancer by exome sequencing.
This is a test.
Merge VarScan 2 Panel
Merge Mpileup files per sample/chromosome into one compressed gzip file per sample.
PreProcess VCF Panel
Preprocess VCF for loading into a annotation database called Gemini. Processes include normalization and decomposition of MNPs by Vt and VCF modification for correct loading into Gemini.
SNP Effect Panel
Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes).
Gemini Annotations Panel
Load functionally annotated VCF file into a MySQL lite annotation database Gemini.
Metrics
Compute metrics and generate coverage tracks per sample. Multiple metrics are computed at this stage: Number of raw reads, Number of filtered reads, Number of aligned reads, Number of duplicate reads, Median, mean and standard deviation of insert sizes of reads after alignment, percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads) whole genome or targeted percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads). A TDF (.tdf) coverage track is also generated at this step for easy visualization of coverage in the IGV browser.
Picard Calculate HS Metrics
Compute on target percent of hybridization based capture.
GATK callable Loci
Computes the callable region or the genome as a bed track.
Extract Common SNP Frequency
Extracts allele frequencies of possible variants across the genome.
BAF Plot
Plots DepthRatio and B allele frequency of previously extracted alleles.
Raw Mpileup
Full pileup (optional). A raw Mpileup file is created using samtools Mpileup and compressed in gzipped format. One packaged Mpileup file is created per sample/chromosome.
Paired Var Scan 2
Variant calling and somatic mutation/CNV detection for next-generation sequencing data. Uses VarScan 2 for Somatic mutation and copy number alteration discovery in cancer by exome sequencing VarScan 2 thresholds based on DREAM3 results generated by SC INFO field remove to prevent collision with Samtools output during ensemble.
Merge Var Scan 2
Merge Mpileup files per sample/chromosome into one compressed gzip file per sample.
Paired Mutect2
GATK MuTect2 Overview caller for SNVs and Indels.
Merge Mutect2
Merge SNVs and indels for GATK MuTect2 Overview Replace TUMOR and NORMAL sample names in VCF to the exact tumor/normal sample names Generate a somatic VCF containing only PASS variants
SAM Tools Paired
Samtools caller for SNVs and Indels using version 0.1.19.
Merge Filter Paired SAM Tools
In this step, bcftools is used to merge the raw binary variants files created in the snpAndIndelBCF step. The output of bcftools is fed to varfilter, which does an additional filtering of the variants and transforms the output into the VCF (.vcf) format. One VCF file contain the SNP/INDEL calls for all samples in the experiment. Additional somatic filters are performed to reduce the number of FPs: 1. vcflibs vcfsamplediff tags each variant with <tag>={germline,`somatic`_,`loh`_} to specify the type of variant given the genotype difference between the two samples. 2. bcftools filter is used to retain only variants with CLR>=15 and have STATUS=somatic from VCFsamplediff 3. bcftools filter is used to retain only variants that have STATUS=germline or STATUS=loh from vcfsamplediff
VarDict Paired
In this step, VarDict caller is used for SNVs and Indels. Note: variants are filtered to remove instance where REF == ALT and REF modified to ‘N’ when REF is AUPAC nomenclature
Merge Filter Paired VarDict
The fully merged VCF is filtered using following steps: 1. Retain only variants designated as somatic by VarDict: either StrongSomatic or LikelySomatic 2. Somatics identified in step 1 must have PASS filter.
Ensemble Somatic
Apply Bcbio.variations ensemble approach for GATK MuTect2 Overview, VarDict, Samtools and VarScan 2 calls Filter ensemble calls to retain only calls overlapping 2 or more callers
GATK Variant Annotator Somatic
Add VCF annotations to ensemble VCF: Standard and Somatic annotations.
Merge GATK Variant Annotator Somatic
Merge annotated somatic VCFs.
Compute Cancer Efects Somatic
Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes). Modified arguments to consider paired cancer data.
Combine Tumor Pairs Somatic
Combine numerous ensemble VCFs into one VCF for Gemini annotations.
All Pairs Compute Effects Somatic
Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes). Modified arguments to consider paired cancer data. Applied to all tumor pairs.
Gemini Annotations Somatic
Load functionally annotated VCF file into a MySQL lite annotation database Gemini.
Ensemble Germline Loh
Apply Bcbio.variations ensemble approach for VarDict, Samtools and VarScan 2 calls Filter ensemble calls to retain only calls overlapping 2 or more callers.
GATK Variant Annotator Germline
Add VCF annotations to ensemble VCF: most importantly the AD field.
Merge GATK Variant Annotator Germline
Merge annotated germline and LOH VCFs.
Compute Cancer Effects Germline
Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes). Modified arguments to consider paired cancer data.
Combine Tumor Pairs Germline
Combine numerous ensemble VCFs into one VCF for Gemini annotations.
All Pairs Compute Effects Germline
Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes). Modified arguments to consider paired cancer data. Applied to all tumor pairs.
Gemini Annotations Germline
Load functionally annotated VCF file into a MySQL lite annotation database Gemini.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to the latest pipeline implementation.
MuTect2 Tool for calling somatic SNVs and indels via local assembly of haplotypes - See here.
A three-caller pipeline for variant analysis of cancer whole-exome sequencing data.