DNA Sequencing (High Coverage) Pipeline¶
DNA Sequencing (High Coverage) is another GenPipes pipeline that is optimized for deep coverage samples. It is based on DNA Sequencing Pipeline.
Introduction¶
The DnaSeq High Coverage Pipeline is based on the DNA Sequencing Pipeline and follows the same initial pipeline steps. The difference begins at the Mark Duplicates step. Since this data is high coverage Mark Duplicates step is not run. Recalibration is not run either because typically, these datasets are targeted with amplicons or custom capture which render recalibration useless. Also variant calling is done only using frequency. Bayesian callers are not used because these typically don’t fare well with the high coverage.
Version¶
3.6.2
For latest pipeline implementation details visit README.md.
Usage¶
dnaseq_high_coverage.py [-h] [--help] [-c CONFIG [CONFIG ...]]
[-s STEPS] [-o OUTPUT_DIR]
[-j {pbs,batch,daemon,slurm}] [-f] [--no-json]
[--report] [--clean]
[-l {debug,info,warning,error,critical}]
[--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-r READSETS] [-v]
[-t {mugqic,mpileup,light,sv}]
Optional Arguments
-t {mugqic,mpileup,light, sv}, --type {mugqic,mpileup,light,sv}
DNAseq High Coverage analysis type
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Following instructions are meant to be run on C3G deployed GenPipes on Beluga server. It uses human genome data available at Beluga server. Use the following command on Beluga server to run DNA Sequencing (high coverage) pipeline:
dnaseq_high_coverage.py -c
$MUGQIC_PIPELINES_HOME/pipelines/dnaseq_high_coverage/dnaseq_high_coverage.base.ini
$MUGQIC_PIPELINES_HOME/pipelines/dnaseq_high_coverage/dnaseq_high_coverage.beluga.ini
-j slurm -s 1-15 -g dna_high_cov_commands.sh
bash dna_high_cov_commands.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
Pipeline Schema¶
Figure below shows the schema of the DNA Sequencing (High Coverage) sequencing protocol.
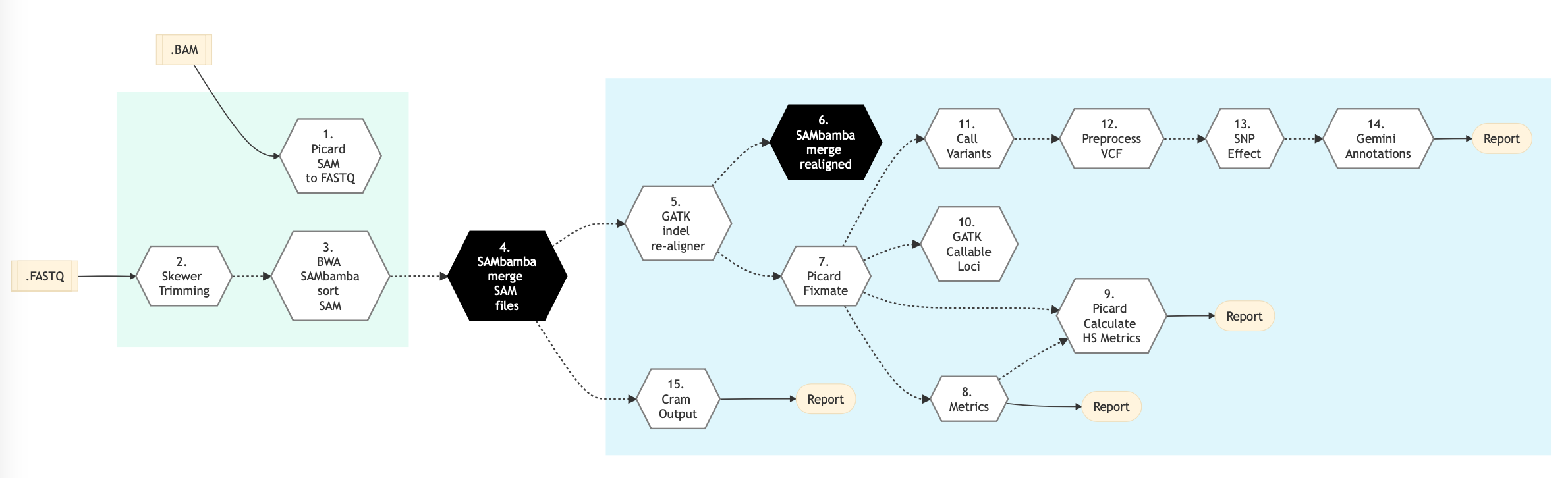
Figure: Schema of DNA Sequencing (High Coverage) Sequencing protocol¶
Pipeline Steps¶
The table below shows various steps that constitute the DNA Sequencing (High Coverage) genomic analysis pipelines.
DNA (High Coverage) sequencing Steps |
|
|---|---|
10 |
|
Step Details¶
Following are the various steps that are part of GenPipes DNA (High Coverage) Sequencing genomic analysis pipeline:
Picard SAM to FASTQ
This step converts SAM/BAM files from the input readset file into FASTQ format if FASTQ files are not already specified in the readset file. Otherwise, it does no processing.
Trimmomatic
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic - Flexible sequencing data trimmer. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Stats
The trim statistics per readset are merged at this step.
GATK Indel Realigner
Insertion and deletion realignment is performed on regions where multiple base mismatches are preferred over indels by the aligner since it can appear to be less costly by the algorithm. Such regions will introduce false positive variant calls which may be filtered out by realigning those regions properly. Realignment is done using GATK Toolkit. The reference genome is divided by a number regions given by the nb_jobs parameter.
Picard Fixmate
After realignment, some read pairs might become unsynchronized. This step verifies that the information is correct between the two pairs and if it is not, it fixes any inaccuracies to avoid problems during the following steps.
Metrics
Compute metrics and generate coverage tracks per sample. Multiple metrics are computed at this stage: Number of raw reads, Number of filtered reads, Number of aligned reads, Number of duplicate reads, Median, mean and standard deviation of insert sizes of reads after alignment, percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads) whole genome or targeted percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads). A TDF (.tdf) coverage track is also generated at this step for easy visualization of coverage in the IGV browser.
Picard Calculate HS Metrics
Compute on target percent of hybridization based capture.
GATK callable loci
Computes the callable region or the genome as a bed track.
Call Variants
This step uses VarScan caller for insertions and deletions.
Pre-process VCF
Pre-process vcf for loading into a annotation database - Gemini. Processes include normalization and decomposition of MNPs by Vt and vcf FORMAT modification for correct loading into Gemini.
SNP Effect
This step performs Variant effect annotation. The .vcf files are annotated for variant effects using the SnpEff software. SnpEff annotates and predicts the effects of variants on genes (such as amino acid changes).
Gemini Annotations
This step loads functionally annotated vcf file into a mysql lite annotation database as part of Gemini processing.
Skewer Trimming
TBD-GenPipes-Dev
BWA SAMbamba Sort SAM
The input for this step is the trimmed FASTQ files if available. Otherwise, it uses the FASTQ files from the readset. If those are not available then it uses FASTQ output files from the previous ‘Picard SAM to FASTQ`_ step where BAM files are converted to FASTQ format.
In this step, filtered reads are aligned to a reference genome. The alignment is done per sequencing readset. The alignment software used is BWA with algorithm: bwa mem. BWA output BAM files are then sorted by coordinate using SAMBAMBA.
SAMBAM Merge SAM Files
This step takes as input files:
Aligned and sorted BAM output files from previous bwa_mem_picard_sort_sam step if available
Else, BAM files from the readset file
In this step, BAM readset files are merged into one file per sample. Merge is done using Picard.
SAMBAM Merge Realigned
In this step, BAM files of regions of realigned reads are merged per sample using SAMBAMBA.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to DNA Sequencing (High Coverage) README.md file.