ChIP Sequencing Pipeline¶
Chromatin Immunoprecipitation (ChIP) sequencing technique is used for mapping DNA-protein interactions. It is a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. The ChIP-Seq workflow is based on the ENCODE Project workflow. It aligns reads using the Burrows-Wheeler Aligner. It creates tag directories using Homer routines. Peaks are called using Model based Analysis for Chip Sequencing (MACS2) and annotated using Homer. Binding motifs are also identified using Homer. Metrics are calculated based on IHEC requirements. The ChIP-Seq pipeline can also be used for assay for transposase-accessible chromatin using sequencing (ATAC-Seq) samples. At GenPipes, we are developing a pipeline that is specific to ATAC-Seq.
Introduction¶
ChIP-Seq experiments allows the isolation and sequencing of genomic DNA bound by a specific transcription factor, covalently modified histone, or other nuclear protein. The pipeline starts by trimming adapters and low quality bases and mapping the reads (single end or paired end ) to a reference genome using Burrows-Wheeler Aligner (BWA). Reads are filtered by mapping quality and duplicate reads are marked. Then, Homer quality control routines are used to provide information and feedback about the quality of the experiment. Peak calls is executed by MACS and annotation and motif discovery for narrow peaks are executed using Homer. Statistics of annotated peaks are produced for narrow peaks and a standard report is generated.
For more details, see ChIP-Seq Guidelines, and MUGQIC_Bioinfo_Chip-Seq.pptx.
Version¶
3.6.2
For the latest implementation and usage details refer to Hi-C Sequencing implementation ChIP-Seq Pipeline README file.
Usage¶
chipseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-d DESIGN] [-t {chipseq, atacseq}] [-r READSETS] [-v]
Optional Arguments
-t {chipseq, atacseq}, --type {chipseq, atacseq}
Type of pipeline (default chipseq)
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Note
ChIPSeq Pipeline Design File Format
Sample MarkName EW22_EW3_vs_EW7_TC71
EW22 H3K27ac 1
EW3 H3K27ac 1
EW7 H3K27ac 2
TC71 H3K27ac 2
where, the numbers specify the sample group membership for this contrast:
'0' or '': the sample does not belong to any group;
'1': the sample belongs to the control group;
'2': the sample belongs to the treatment test case group.
The design file only accepts 1 for control, 2 for treatment and 0 for other samples that do not need to compare.
Warning
Incorrect Design File Format
Please note that the first and second column in the design file must be sample name and histone mark/binding protein respectively.
If the user specifies any value other than the permitted ones above in the design file, the pipeline will fail.
Example Run¶
You can download ChIP-Seq test dataset and use the following command to execute the ChIP-Seq genomics pipeline:
chipseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/chipseq/chipseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/chipseq/chipseq.beluga.ini -r readset.chipseq.txt -d design.chipseq.txt -s 1-20 -g chipseqScript.txt
bash chipseqScript.txt
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
The commands will be sent to the job queue and you will be notified once each step is done. If everything runs smoothly, you should get MUGQICexitStatus:0 or Exit_status=0. If that is not the case, then an error has occurred after which the pipeline usually aborts. To examine the errors, check the content of the job_output folder.
Pipeline Schema¶
Figure below shows the schema of ChIP-Seq protocol.
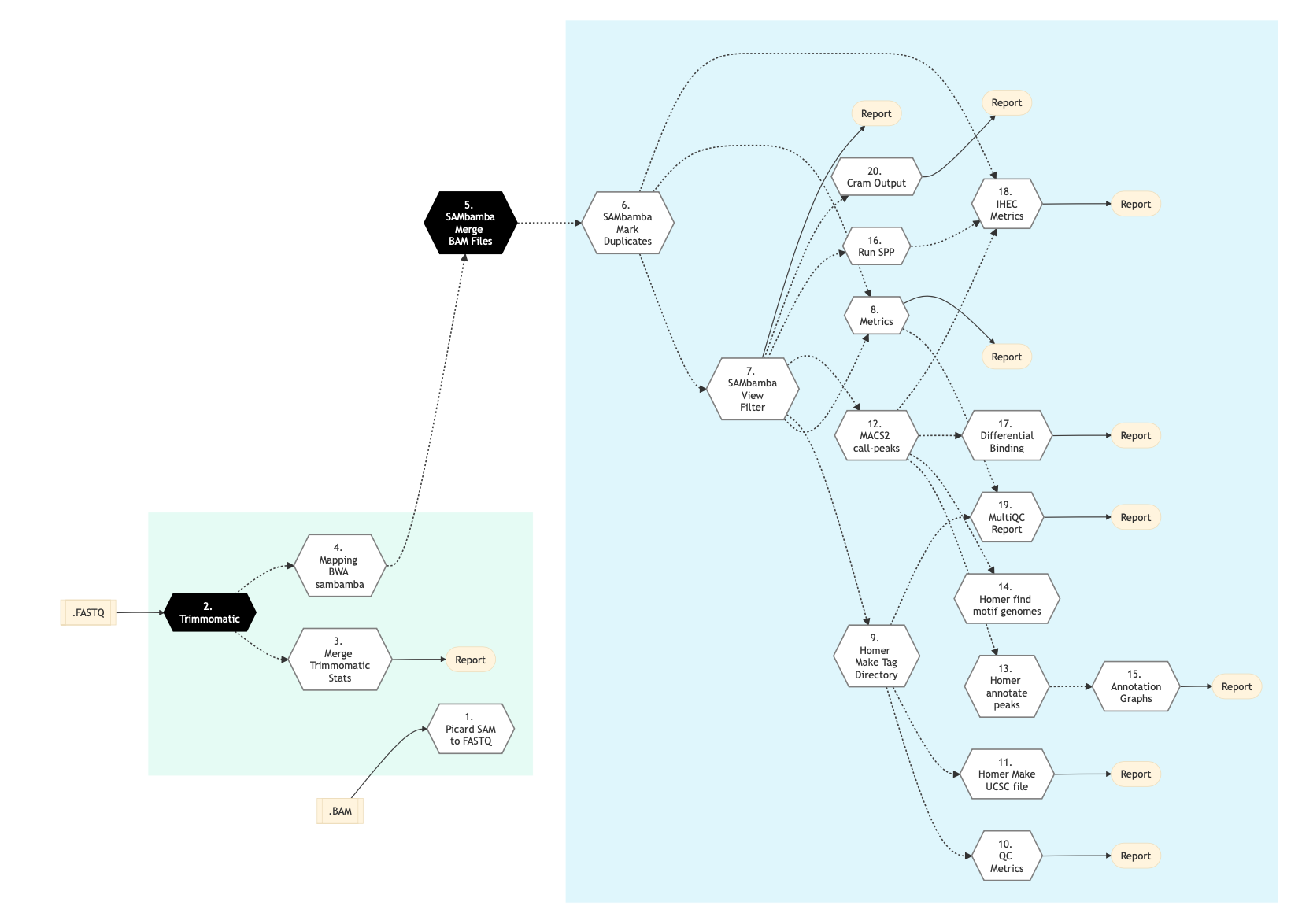
Figure: Schema of ChIP Sequencing protocol¶
Following is the schema for the ChIP-Seq pipeline using the -t atacseq option:
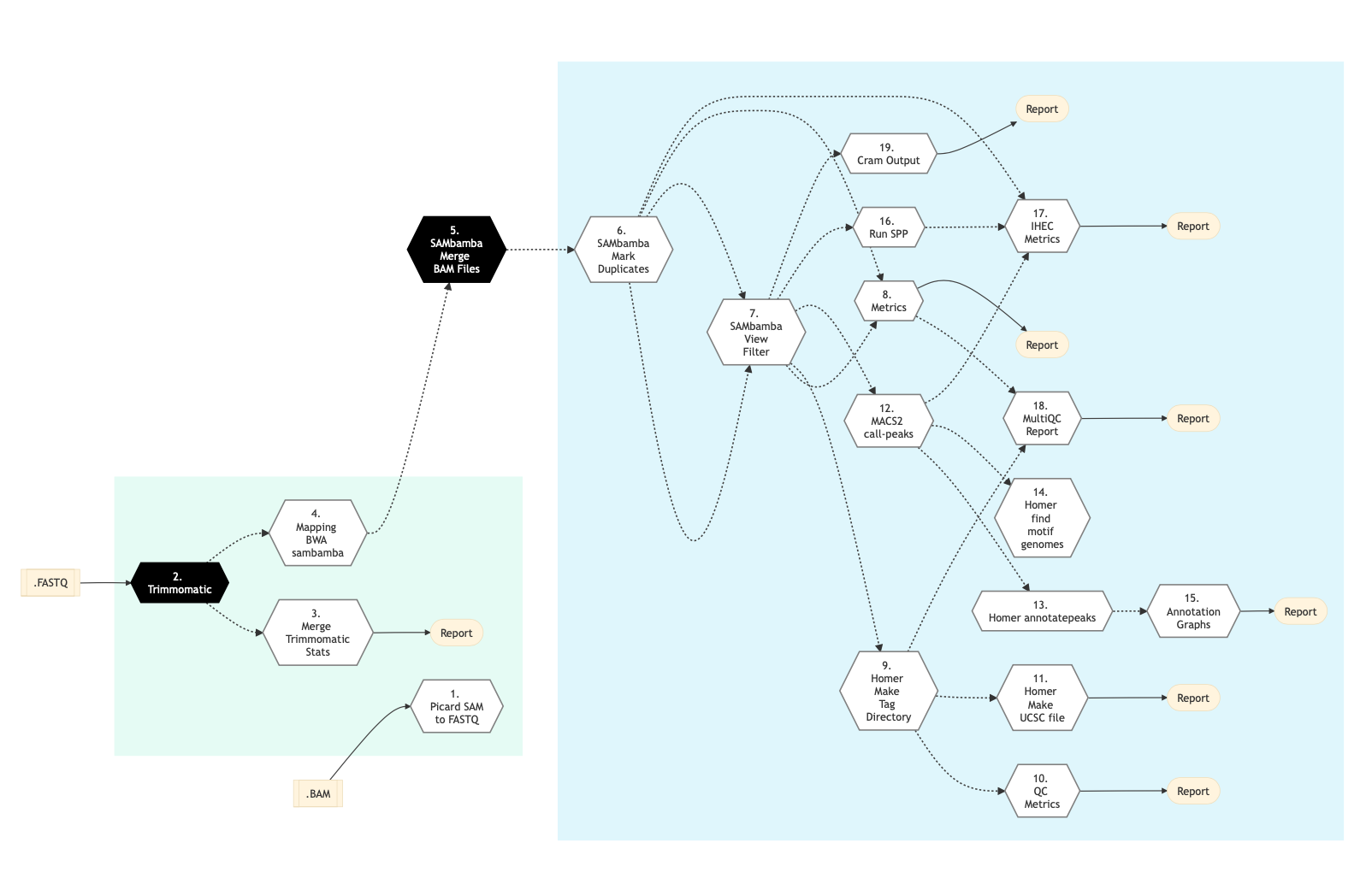
Figure: Schema of ChIP Sequencing -t atacseq protocol¶
Pipeline Steps¶
The table below shows various steps that constitute the ChIP sequencing pipeline:
ChIP Sequencing Steps |
ChIP Sequencing (atacseq) |
|
|---|---|---|
Step Details¶
Following are the various steps that are part of GenPipes ChIP-Seq genomic analysis pipeline:
Picard Sam to Fastq
If FASTQ files are not already specified in the Readset file, then this step converts SAM/BAM files from the input Readset into FASTQ format. Otherwise, it does nothing.
Trimmomatic
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic Process. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
If available, trimmomatic step in Hi-C analysis takes FASTQ files from the readset file as input. Otherwise, it uses the FASTQ output file generated from the previous Picard Sam to Fastq step conversion of the BAM files.
Merge Trimmomatic Stats
The trim statistics per Readset file are merged at this step.
Mapping BWA Mem Sambamba
This step takes as input files trimmed FASTQ files, if available. Otherwise it takes FASTQ files from the readset. If readset is not supplied then it uses FASTQ output files from the previous Picard Sam to Fastq conversion of BAM files.
SAMbamba Merge BAM
BAM readset files are merged into one file per sample. Merge is done using Sambamba.
If available, the aligned and sorted BAM output files from previous Mapping BWA Mem Sambamba step are used as input. Otherwise, BAM files from the readset file is used as input.
SAMbamba Mark Duplicates
Mark duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be marked as a duplicate in the BAM file. Marking duplicates is done using SAMbamba.
SAMbamba View Filter
Filter unique reads by mapping quality using SAMbamba.
Metrics
The number of raw/filtered and aligned reads per sample are computed at this stage.
Homer Make Tag Directory
The Homer Tag directories, used to check for quality metrics, are computed at this step.
QC Metrics
Sequencing quality metrics as tag count, tag auto-correlation, sequence bias and GC bias are generated.
Homer Make UCSC files
Wiggle Track Format files are generated from the aligned reads using Homer. The resulting files can be loaded in browsers like IGV or UCSC.
MACS2 call peak
Peaks are called using the MACS2 software. Different calling strategies are used for narrow and broad peaks. The mfold parameter used in the model building step is estimated from a peak enrichment diagnosis run. The estimated mfold lower bound is 10 and the estimated upper bound can vary between 15 and 100.
The default mfold parameter of MACS2 is [10,30].
MACS2 ATAC-seq call peak
Assay for Transposon Accessible Chromatin (ATAC-seq) analysis enables measurement of chromatin structure modifications (nucleosome free regions) on gene regulation. This step involves calling peaks using the MACS2 software. Different calling strategies are used for narrow and broad peaks. The mfold parameter used in the model building step is estimated from a peak enrichment diagnosis run. The estimated mfold lower bound is 10 and the estimated upper bound can vary between 15 and 100.
The default mfold parameter of MACS2 is [10,30].
Homer annotate peaks
The peaks called previously are annotated with HOMER tool using RefSeq annotations for the reference genome. Gene ontology and genome ontology analysis are also performed at this stage.
Homer find motifs genome
In this step, De novo and known motif analysis per design are performed using HOMER.
Annotation Graphs
This step focuses on peak location statistics. The following peak location statistics are generated per design: proportions of the genomic locations of the peaks. The locations are: Gene (exon or intron), Proximal ([0;2] kb upstream of a transcription start site), Distal ([2;10] kb upstream of a transcription start site), 5d ([10;100] kb upstream of a transcription start site), Gene desert (>= 100 kb upstream or downstream of a transcription start site), Other (anything not included in the above categories); The distribution of peaks found within exons and introns; The distribution of peak distance relative to the transcription start sites (TSS); the Location of peaks per design.
Run SPP
This step runs spp to estimate NSC and RSC ENCODE metrics. For more information - see quality enrichment of ChIP sequence data, phantompeakqualtools.
Differential Binding
Differential Binding step is meant for processing DNA data enriched for genomic loci, including ChIP- seq data enriched for sites where specific protein binding occurs, or histone marks are enriched. It uses DiffBind package that helps in identifying sites that are differentially bound between sample groups.
GenPipes Chipseq pipeline performs differential binding based on the provided treatments and controls as per the particular comparison specified in the design file. The differential analysis results are generated separately for each specified comparison in the Design File, with correctly specified treatments (2) and controls (1) samples.
Samples with ‘0’ (zero) are ignored during the comparison. For details regarding how to specify sample group membership in the design file, refer to Design File Format details.
For comparison, at least two samples for each group must be specified. If two samples per group are not specified, the differential binding step will be skipped during the pipeline run.
Warning
Incorrect Design File Format
If the specified design file does not follow the specified Design File Format for ChIPseq pipeline, the differential binding step will be skipped during the pipeline run.
The output of differential analysis containing differentially bound peaks are saved as a TSV. In addition, for each comparison done using DiffBind, an html report is also generated for QC differential analysis.
IHEC Metrics
This step generates IHEC’s standard metrics.
MultiQC Report
A quality control report for all samples is generated. For more detailed information see MultiQC documentation.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More Information¶
For the latest implementation and usage details, see ChIP-Seq Pipeline README. Here is some more information about ChIP sequencing pipeline that you may find interesting.
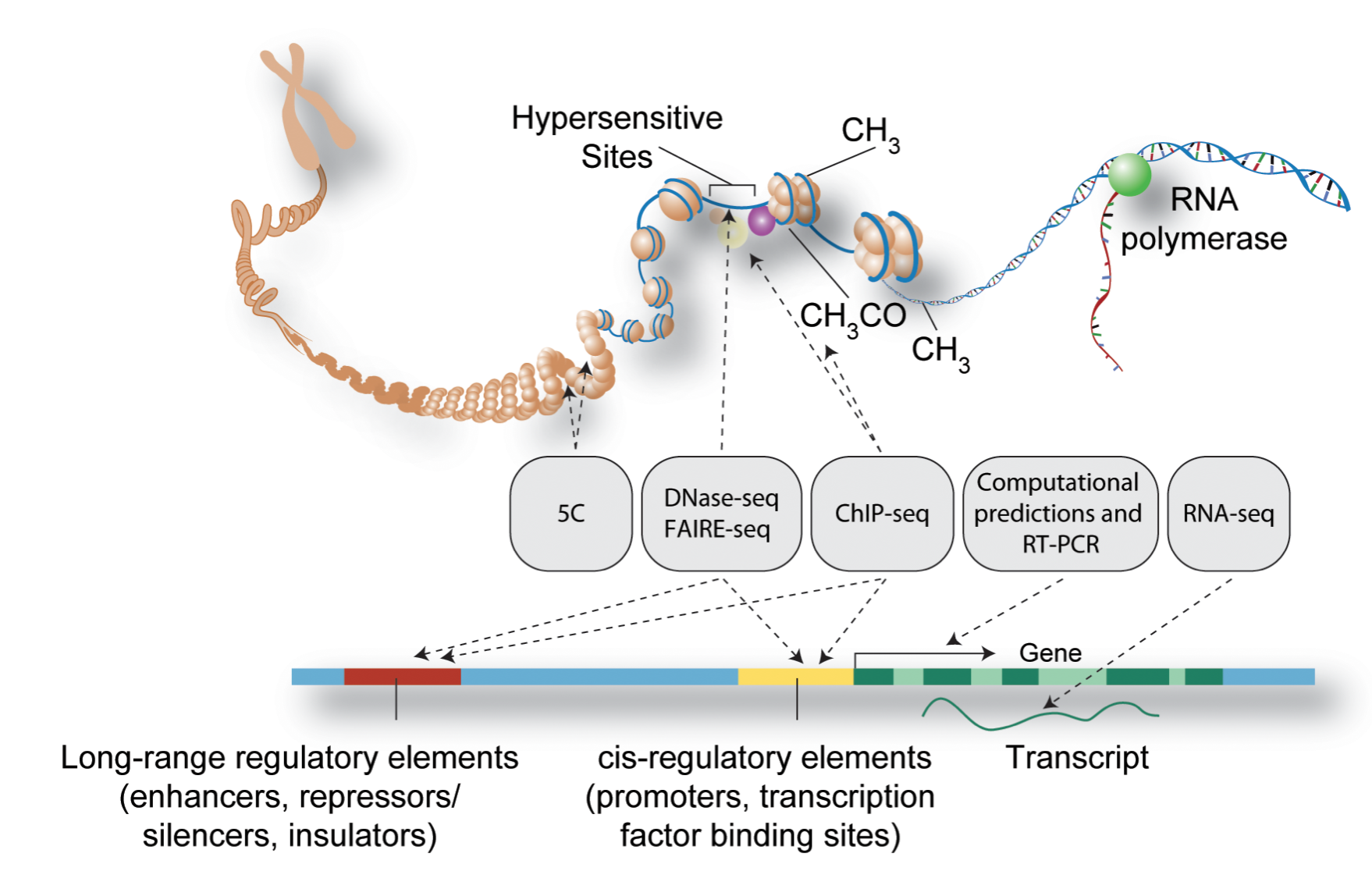
Figure: Schematic representation of major methods used to detect functional elements in DNA (Source PLOS)¶