RNA Sequencing Pipeline¶
This pipeline aligns reads with STAR 2-passes mode, assembles transcripts with Cufflinks, and performs differential expression with Cuffdiff. In parallel, gene-level expression is quantified using htseq-count, which produces raw read counts that are subsequently used for differential gene expression with both DESeq process and edgeR algorithms. Several common quality metrics (e.g., ribosomal RNA content, expression saturation estimation) are also calculated through the use of RNA-SeQC and in-house scripts. Gene Ontology terms are also tested for over-representation using GOseq. Expressed short single-nucleotide variants (SNVs) and indels calling is also performed by this pipeline, which optimizes GATK best practices to reach a sensitivity of 92.8%, precision of 87.7%, and F1 score of 90.1%.
Introduction¶
The standard MUGQIC RNA-Seq pipeline is based on the use of the STAR aligner to align reads to the reference genome. These alignments are used during downstream analysis to determine genes and transcripts differential expression. The Cufflinks suite is used for the transcript analysis whereas DESeq and edgeR are used for the gene analysis. The RNAseq pipeline requires to provide a design file which will be used to define group comparison in the differential analyses. The differential gene analysis is followed by a Gene Ontology (GO) enrichment analysis. This analysis use the goseq approach. The goseq is based on the use of non-native GO terms (see details in the section 5 of the corresponding vignette.
Finally, a summary html report is automatically generated by the pipeline at the end of the analysis. This report contains description of the sequencing experiment as well as a detailed presentation of the pipeline steps and results. Various Quality Control (QC) summary statistics are included in the report and additional QC analysis is accessible for download directly through the report. The report includes also the main references of the software tools and methods used during the analysis, together with the full list of parameters that have been passed to the pipeline main script.
See More information section below for details.
Version¶
3.6.2
For the latest implementation and usage details refer to RNA Sequencing implementation README file file.
Usage¶
usage: rnaseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-d DESIGN]
[-t {cufflinks,stringtie}] [-r READSETS] [-v]
Optional Arguments
-t {cufflinks,stringtie}, --type {cufflinks,stringtie}
Type of RNA-seq assembly method (default stringtie,
faster than cufflinks)
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
You can download RNA Sequencing Pipeline test dataset and use the following command to execute the RNA Sequencing genomics pipeline:
rnaseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/rnaseq/rnaseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/rnaseq/rnaseq.cedar.ini workshop.ini -r readset.rnaseq.txt -d design.rnaseq.txt -s 1-24 -j slurm -g commands.txt
bash commands.txt
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
This set of commands is meant for running GenPipes on C3 data center. For more details, you can refer to GenPipes RNA Sequencing Workshop 2018 presentation.
Pipeline Schema¶
Figure below shows the schema of the RNA sequencing protocol (cufflinks).
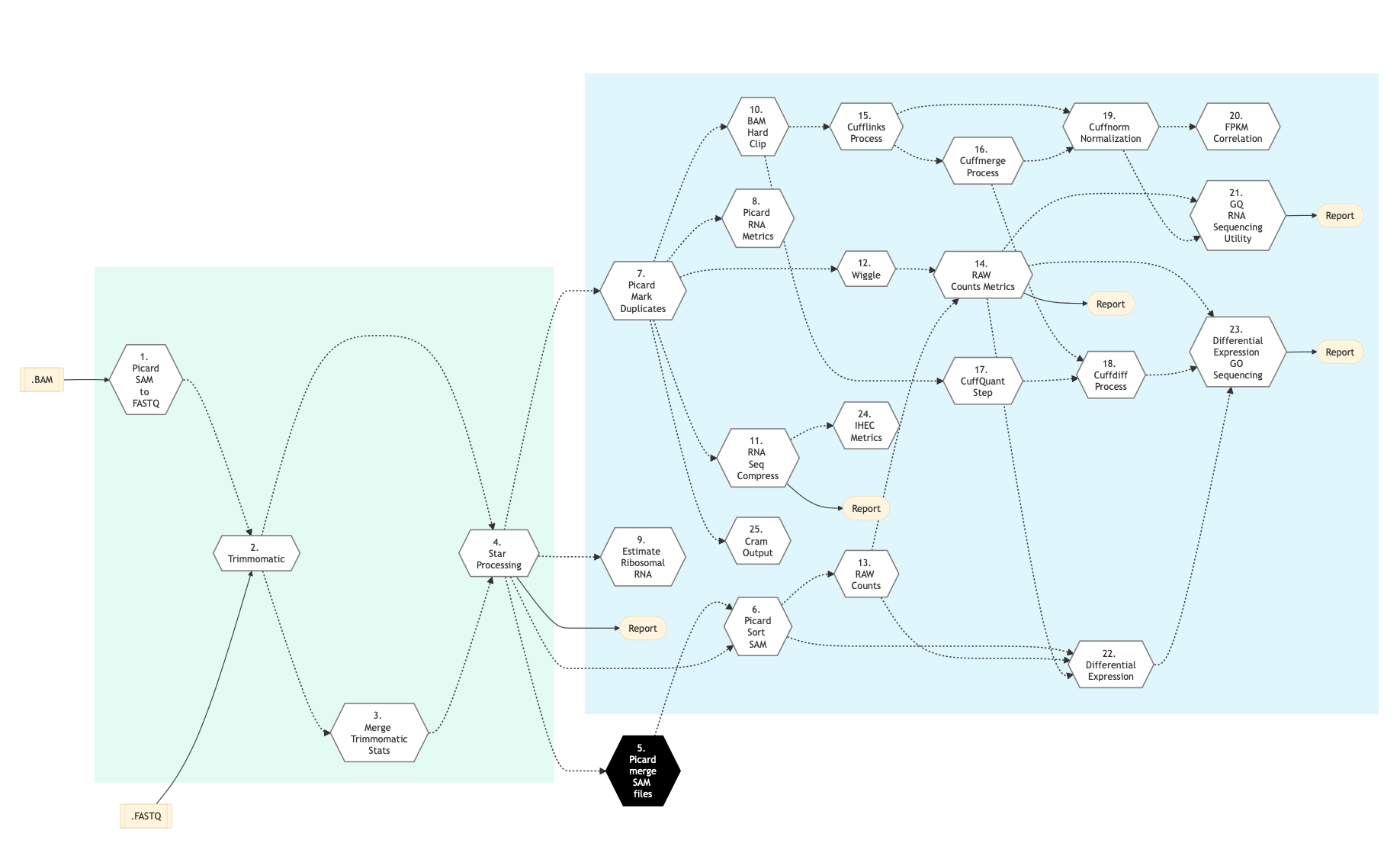
Figure: Schema of RNA Sequencing pipeline (Cufflinks)¶
The following figure shows the schema of the RNA sequencing protocol (stringtie).
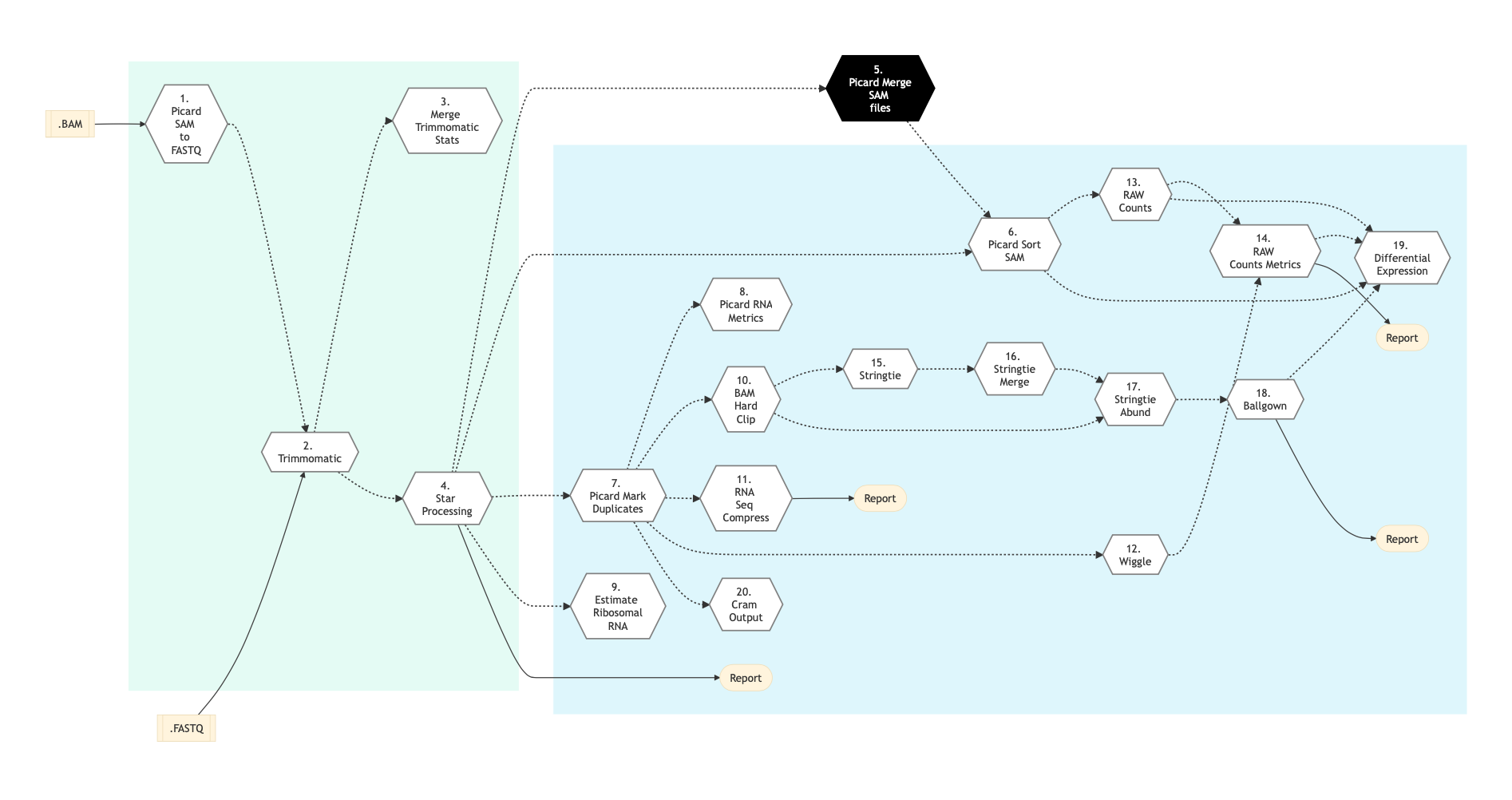
Figure: Schema of RNA Sequencing pipeline (Stringtie)¶
Pipeline Steps¶
The table below lists various steps of GenPipes RNA Sequencing Pipeline:
RNA Sequencing (Cufflinks) |
RNA Sequencing (Stringtie) |
|
|---|---|---|
Step Details¶
Picard SAM to FastQ
In this step, if FASTQ files are not already specified in the readset file, then it converts SAM/BAM files from the input readset file into FASTQ format. Otherwise, nothing is done.
Trimmomatic
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic tool. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Stats
The trim statistics per readset are merged at this step.
Star Processing
The filtered reads are aligned to a reference genome. The alignment is done per readset of sequencing using the STAR software. It generates a Binary Alignment Map file (.bam).
This step takes as input files:
Trimmed FASTQ files if available
Else, FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Picard Merge SAM Files
BAM readset files are merged into one file per sample. Merge is done using Picard Tool.
Picard Sort SAM
The alignment file is reordered (QueryName) using Picard Tool. The QueryName-sorted BAM files will be used to determine raw read counts.
Picard Mark Duplicates
Mark duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be marked as a duplicate in the BAM file. Marking duplicates is done using Picard package.
Picard RNA Metrics
Computes a series of quality control metrics using both CollectRnaSeqMetrics and CollectAlignmentSummaryMetrics functions metrics are collected using Picard package.
Estimate Ribosomal RNA
This step uses readset BAM files and bwa mem to align reads on the rRNA reference fasta and count the number of read mapped The filtered reads are aligned to a reference fasta file of ribosomal sequence. The alignment is done per sequencing readset. The alignment software used is BWA package with algorithm: bwa mem. BWA output BAM files are then sorted by coordinate using Picard package.
BAM Hard Clip
Generate a hardclipped version of the BAM for the toxedo suite which does not support this official SAM feature.
RNA Seq Compress
Computes a series of quality control metrics using RNA SeQC processing.
Wiggle
Generate wiggle tracks suitable for multiple browsers.
Raw Counts
Count reads in feature using HT Seq Count.
Raw Counts Metrics
Create rawcount matrix, zip the wiggle tracks and create the saturation plots based on standardized read counts.
Cufflinks Process
Compute RNA-Seq data expression using cufflinks package. Warning: It needs to use a hard clipped bam file while Tuxedo tools do not support official soft clip SAM format.
Cuffmerge Process
Merge assemblies into a master transcriptome reference using cuffmerge package.
Cuffquant Step
Compute expression profiles (abundances.cxb) using cuffquant. Warning: It needs to use a hard clipped bam file while Tuxedo tools do not support official soft clip SAM format.
Cuffdiff Process
Cuffdiff package is used to calculate differential transcript expression levels and test them for significant differences.
Cuffnorm Normalization
This step performs global normalization of RNA-Sequence expression levels using Cuffnorm algorithm.
FPKM Correlation
Compute the pearson correlation matrix of gene and transcripts FPKM. FPKM data are those estimated by cuffnorm.
GQ RNA Sequencing Utility
Exploratory analysis using the gqSeqUtils R package.
Differential Expression
Performs differential gene expression analysis using DESEQ package and EDGER package. Merge the results of the analysis in a single csv file.
Differential Expression GO sequencing
Gene Ontology analysis for RNA-Seq using the Bioconductor’s R package goseq. Generates GO annotations for differential gene expression analysis.
IHEC Metrics
Generate IHEC’s standard metrics.
Stringtie
Assemble transcriptome using Stringtie assembler.
Stringtie Assemble Transcriptome
This step assembles transcriptome and compute RNA-seq expression using Stringtie assembler.
Stringtie Merge
Merge assemblies into a master transcriptome reference using Stringtie assembler.
Ballgown Gene Expression
Ballgown tool is used to calculate differential transcript and gene expression levels and test them for significant differences.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to README.md.