HiC Sequencing Pipeline¶
The HiC-Seq workflow aligns reads using HiCUP. It creates tag directories, produces interaction matrices, and identifies compartments and significant interactions using Homer. It identifies topologically associating domains using TopDom and RobusTAD (bioRxiv 293175). It also creates “.hic” files using JuiceBox and metrics reports using MultiQC. The HiC-Seq workflow can also process capture Hi-C data with the flag “-t capture” using CHICAGO.
Introduction¶
Hi-C experiments allow researchers to understand chromosomal folding and structure using proximity ligation techniques. This pipeline analyzes both Hi-C experimental data (-t hic) and capture Hi-C data (-t capture).
Hi-C
The Hi-C pipeline, selected using the “-t hic” parameter, starts by trimming adapters and low quality bases. It then maps the reads to a reference genome using HiCUP. HiCUP first truncates chimeric reads, maps reads to the genome, then it filters Hi-C artifacts and removes read duplicates. Samples from different lanes are merged and a tag directory is created by Homer, which is also used to produce the interaction matrices and compartments. TopDom is used to predict topologically associating domains (TADs) and Homer is used to identify significant interactions.
Hi-C capture
The capture Hi-C pipeline, selected using the “-t capture” parameter, starts by trimming adapters and low quality bases. It then maps the reads to a reference genome using HiCUP. HiCUP first truncates chimeric reads, maps reads to the genome, then it filters Hi-C artifacts and removes read duplicates. Samples from different lanes are merged and CHiCAGO is then used to filter capture-specific artifacts and call significant interactions. This pipeline also identifies enrichment of regulatory features when provided with ChIP-Seq marks. It can also return bed interactions with significant baits (bait_intersect step) or with captured interactions (capture_intersect step).
An example of the Hi-C report for an analysis on public data (GM12878 Rao. et al.) is available for illustration purposes only: Hi-C report.
Version¶
3.6.2
For the latest implementation and usage details refer to Hi-C Sequencing implementation README.md file.
Usage¶
hicseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
-e {DpnII,HindIII,NcoI,MboI,Arima} [-t {hic,capture}]
[-r READSETS] [-v]
Optional Arguments
-e {DpnII,HindIII,NcoI,MboI,Arima},
--enzyme {DpnII,HindIII,NcoI,MboI,Arima}
Restriction Enzyme used to generate Hi-C library (default DpnII)
-t {hic,capture},
--type {hic,capture} Hi-C experiment type (default hic)
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
You can download Hi-C test dataset and run the following commands:
hicseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/hicseq/hicseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/hicseq/hicseq.guillimn.ini -r readsets.HiC010.tsv -s 1-15 -e MboI -j pbs -g hicseqScript_SRR1658581.txt
bash hicseqScript_SRR1658581.txt
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
Pipeline Schema¶
Figure below shows the schema of the Hi-C protocol.
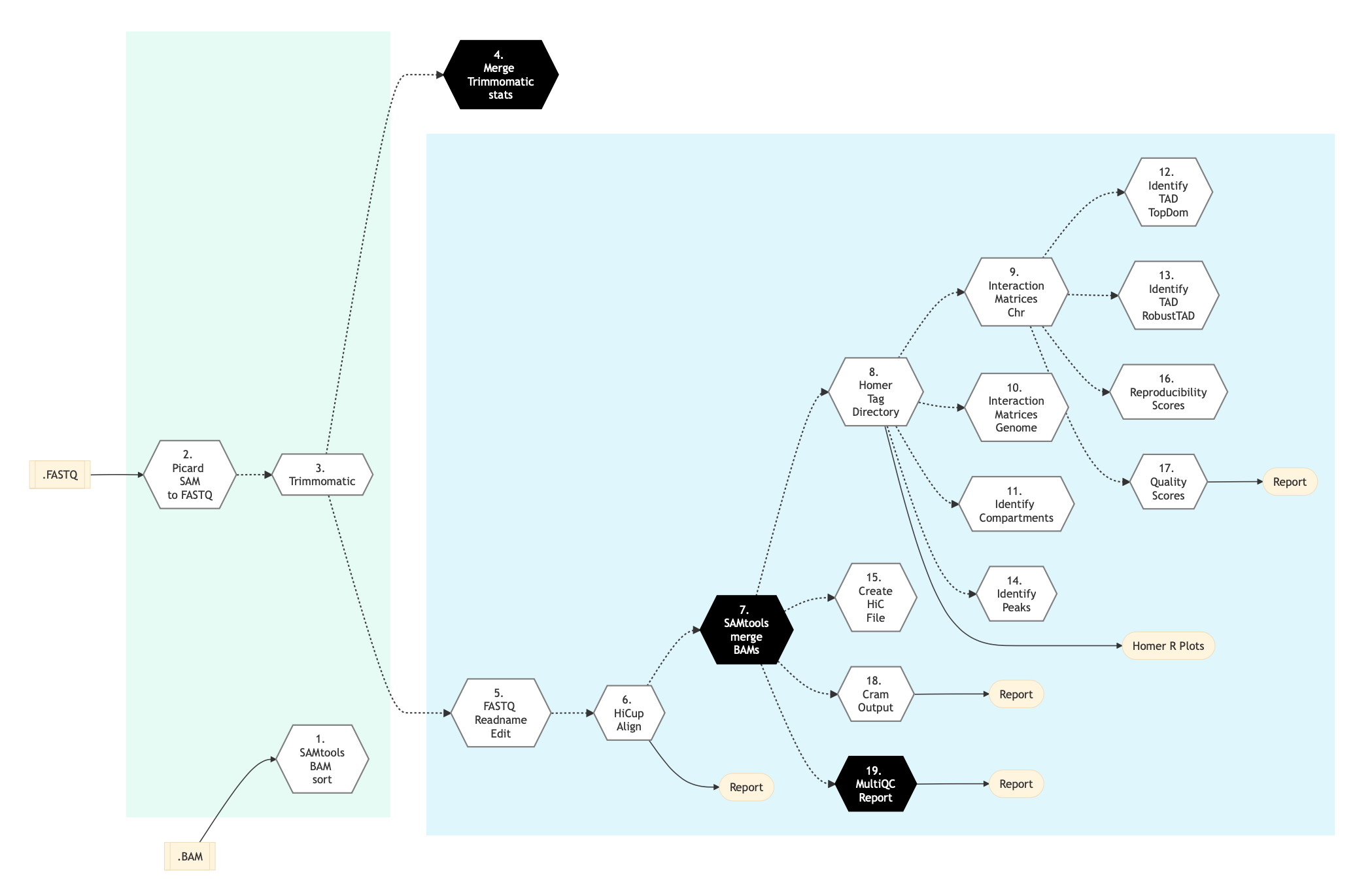
Figure: Schema of Hi-C Sequencing protocol¶
The following figure shows the pipeline schema for capture Hi-C protocol.
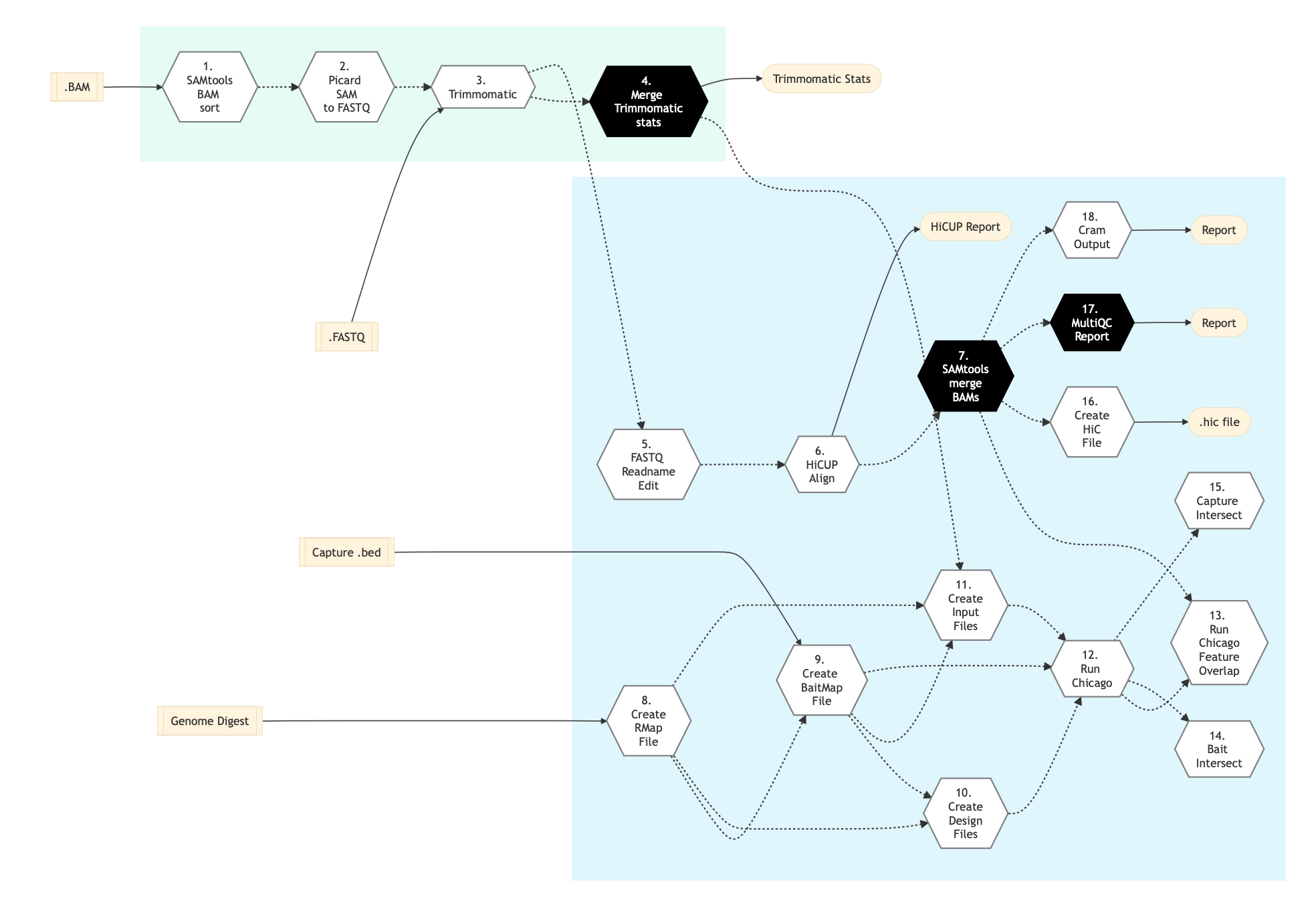
Figure: Schema of Hi-C capture protocol¶
The following figure shows the pipeline schema for capture Hi-C protocol.
Pipeline Steps¶
The table below shows various steps that constitute the Hi-C and Hi-C capture genomic analysis pipelines.
Hi-C sequencing Steps |
Hi-C Capture sequencing Steps |
|
|---|---|---|
Step Details¶
Following are the various steps that are part of Hi-C genomic analysis pipelines:
Samtools Bam Sort
Sorts BAM files by readname prior to Picard Sam to Fastq step, in order to minimize memory consumption. If BAM file is small and the memory requirements are reasonable, this step can be skipped.
Picard Sam to Fastq
If FASTQ files are not already specified in the Readset file, then this step converts SAM / BAM files from the input Readset into FASTQ format. Otherwise, it does nothing.
Trimmomatic
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic Process. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
If available, trimmomatic step in Hi-C analysis takes FASTQ files from the readset file as input. Otherwise, it uses the FASTQ output file generated from the previous Picard Sam to Fastq step conversion of the BAM files.
Merge Trimmomatic Stats
The trim statistics per Readset file are merged at this step.
Fastq ReadName Edit
Removes the added /1 and /2 by Picard Sam to Fastq transformation to avoid issues with downstream software like Homer.
HiCUP Align
Paired-end Hi-C reads are truncated, mapped and filtered using HiCUP. The resulting BAM file is filtered for Hi-C artifacts and duplicated reads. It is ready for use as input for downstream analysis. For more detailed information about the HiCUP process visit HiCUP Project Page.
Samtools Merge Bams
BAM readset files are merged into one file per sample. Merge is done using samtools. This step takes as input files the aligned BAM / SAM files from the Hicup Align step.
Homer Tag Directory
The BAM file produced by HiCUP is used to create a tag directory using Homer for further analysis that includes interaction matrix generation, compartments and identifying significant interactions. For more details, visit Homer.
Interaction Matrices Chr
In this step, intra-chromosomal interaction matrices are produced by Homer Matrices at resolutions defined in the ini config file and plotted by HiCPlotter.
Interaction Matrices Genome
Genome-wide interaction matrices are produced by Homer at resolutions defined in the ini config file. See Homer Matrices for details.
Identify Compartments
Genomic compartments are identified using Homer at resolutions defined in the ini config file. For details, see Homer Compartments.
Identify TADs TopDom
Topological Associating Domains (TADs) are identified using TopDom at resolutions defined in the ini config file. For details, see TopDom.
Identify TADs RobusTAD
In this step, topological associating Domain (TAD) scores are calculated using RobusTAD for every bin in the genome. RobusTAD is resolution-independent and will use the first resolution in “resolution_TADs” under [identify_TADs] in the ini file. For details, see latest README.md for RobusTAD.
Identify Peaks
This step users Homer Peaks to identify significant intra-Chromosomal interactions or peaks.
Create HiC File
In this step, a .hic file is created per sample in order to visualize in JuiceBox , WashU epigenome browser or as input for other tools.
Create Baitmap File
Here, the baitmap file for Chicago capture analysis is created using the created RMAP file and the probe capture bed file.
Create Design Files
In this step, the design files (NPerBin file (.npb), nbaitsperbin file (.nbpb), Proximal Other End (proxOE) file (.poe)) for Chicago capture analysis are created using the RMAP file and the baitmap file.
Create Input Files
Here, the input file (sample.chinput) for Chicago capture analysis is created using the RMAP file, the baitmap file and the hicup aligned BAM.
Run Chicago
Chicago is run on capture data. Chicago will filter capture hic artifacts and identify significant interactions. It will output data as a bed file and will also output SeqMonk and WashU tracks. For more detailed information about the Chicago, including how to interpret the plots, visit Chicago documentation.
RunChicago FeatureOverlap
This step runs the feature enrichment of Chicago significant interactions. See Chicago documentation for details.
Bait Intersect
With a bed file as input, for example a bed of GWAS SNPs or features of interest, this method returns the lines in the bed file that intersect with the baits that have significant interactions. Input bed must have 4 columns (<chr> <start> <end> <annotation>) and must be tab separated.
Capture Intersect
With bed file as input, for example a bed of GWAS SNPs or features of interest, this method returns the lines in the bed file that intersect with the captured ends (“Other Ends”) that have significant interactions. Input bed must have 4 columns (<chr> <start> <end> <annotation>) and must be tab separated.
Create RMAP File
A RMAP file for Chicago Capture Analysis is created using the HiCUP digestion file.
Multiqc Report
A quality control report for all samples is generated. For more detailed information see MultiQC documentation.
Reproducibility Scores
In this step, R package hicrep is used for calculating the inter-chromosomal reproducibility score. Hicrep considers spatial features in Hi-C data, such as domain structure and distance-dependence, so that there are no misleading results in Hi-C data reproducibility while studying genome-wide chromatin interactions.
The novel reproducibility measure in hicrep can assess pairwiseß differences between Hi-C matrices under a wide range of settings, and can be used to determine optimal sequencing depth. Compared to existing approaches, it consistently shows higher accuracy in distinguishing subtle differences in reproducibility and depicting interrelationships of cell lineages than existing approaches.
Pairwise reproducibility scores are calculated for each chromosome pair in each sample pair, using hicrep at resolutions (bin size) defined in Interaction Matrices Chr step. Other parameters are defined in reproducibility_scores section of the hicseq.base.ini file.
All the scores are finally merged together and an output .csv file is created using the parameters specified during the analysis. Chromosome number, reproducibility scores, standard deviation and smoothing value are used in the analysis. In order to compare samples, ensure that the smoothing value and the sequencing depth are similar across samples.
Down sampling of samples can be performed using the down_sampling parameter in the .ini config file. Correlation matrices and wight matrices can be saved using the following initialization values in the .ini config file:
corr=TRUE
weights=TRUE
Quality Scores
In this step, Quality score per chromosome for each sample is calculated using QuSAR-QC at all resolutions, sequencing depth (coverage) and down_sampling value (coverage) defined in quality_scores step of .ini config file. QUSAR-QC is a part of the hifive hic-seq analysis suite.
Quality Assessment of Spatial Arrangement Reproducibility (QuASAR) transformation and scoring uses the consistency between the raw interaction matrix and the correlation matrix of distance-corrected signal to determine sample quality. For all interactions less than or equal to 100 times the resolution of size, the correlation value is calculated.
In order to determine the quality score for a given chromosome, the weighted average of the correlation values (weighted by the raw interaction signal) minus the unweighted correlation signal average is calculated. A genome-wide value is calculated by summing all numerators and denominators across all chromosomes prior to dividing and subtracting score components. The replicate score is calculated by finding the correlation between the weighted correlation matrices for two samples.
For details, see hifive suite.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to Hi-C Sequencing implementation README.md file.
Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome - Paper introducing Hi-C.
A high-resolution map of the three-dimensional chromatin interactome in human cells. Defining target gene using Hi-C.