Methylation Sequencing Pipeline¶
DNA Methylation is an important epigenetic system related to gene expression. It is involved in embryonic development, genomic imprinting, X chromosome inactivation and cell differentiation. Since methylation takes part in many normal cellular functions, aberrant methylation of DNA may be associated with a wide spectrum of human diseases, including cancer. Bisulfite sequencing approaches are currently considered a “gold standard” allowing comprehensive understanding of the methylome landscape. Whole-genome bisulfite sequencing (WGBS), provides single-base resolution of methylated cytosines across the entire genome.
GenPipes methylation sequencing workflow is adapted from the Bismark pipeline. It aligns paired-end reads with Bowtie 2 default mode. Duplicates are removed with Picard, and methylation calls are extracted using Bismark. Wiggle tracks for both read coverage and methylation profile are generated for visualization. Variant calls can be extracted from the whole-genome bisulfite sequencing (WGBS) data directly using Bis-SNP caller. Bisulfite conversion rates are estimated with lambda genome or from human non-CpG methylation directly. Several metrics based on IHEC requirements are also calculated. Methylation sequencing can also process capture data if provided with a capture BED file.
Both paired-end reads as well as single-end reads are supported by this pipeline. Paired-end reading improves the ability to identify the relative positions of various reads in the genome making it much more effective than single-end reading for resolving structural rearrangements such as gene insertions, deletions or inversions and assembly of repetitive regions. Single-end reads are much less expensive in terms of resource consumption and time needed for analysis and can be used for experiments that do not require higher degrees of accuracy offered by paired-reads.
Introduction¶
The standard MUGQIC Methyl-Seq pipeline uses Bismark to align reads to the reference genome. Treatment and filtering of mapped reads approaches as mark duplicate reads, recalibration and sort are executed using Picard and GATK. Samtools MPILEUP and BCFtool are used to produce the standard SNP and indels variants file (VCF). Additional SVN annotations mostly applicable to human samples include mappability flags, dbSNP annotation and extra information about SVN by using published databases. The SNPeff tool is used to annotate variants using an integrated database of functional predictions from multiple algorithms (SIFT, Polyphen 2, LRT and MutationTaster, PhyloP and GERP++, etc.) and to calculate the effects they produce on known genes.
A summary html report is automatically generated by the pipeline. This report contains description of the sequencing experiment as well as a detailed presentation of the pipeline steps and results. Various Quality Control (QC) summary statistics are included in the report and additional QC analysis is accessible for download directly through the report. The report includes also the main references of the software and methods used during the analysis, together with the full list of parameters that have been passed to the pipeline main script.
Version¶
3.6.2
For the latest implementation and usage details refer to WGS or Methylation Sequencing implementation README file file.
Usage¶
methylseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>
[--genpipes_file GENPIPES_FILE]
[-d DESIGN] [-r READSETS]
[-v]
Optional Arguments
-t {mugqic,mpileup,light, sv}, --type {mugqic,mpileup,light,sv}
Methylseq analysis type
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run¶
Use the following commands to execute the methylation pipeline:
methylseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/methylseq/methylseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/methylseq/methylseq.guillimin.ini -r readset.methylseq.txt -s 1-14 -g methylseq.sh
bash methylseq.sh
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].guillimin.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline in Reference section.
Pipeline Schema¶
Figure below shows the schema of the WGS or Methylation sequencing pipeline.
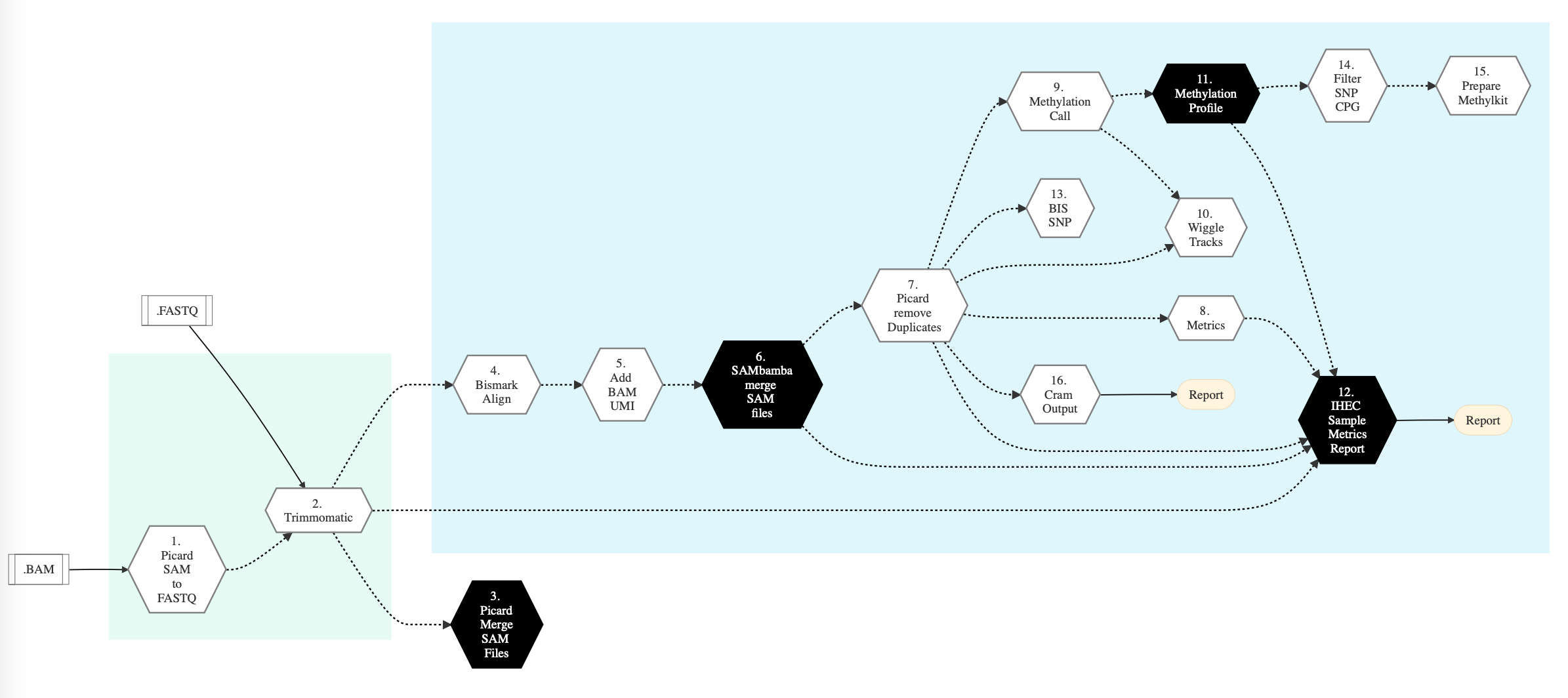
Figure: Schema of WGS or Methylation Sequencing pipeline¶
Pipeline Steps¶
The table below shows various steps that constitute the WGS or Methylation Sequencing genomic analysis pipelines.
Methylation sequencing Steps |
|
|---|---|
Step Details¶
Picard SAM to FastQ
In this step, if FASTQ files are not already specified in the readset file, then it converts SAM/BAM files from the input readset file into FASTQ format. Otherwise, nothing is done.
Trimmomatic
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic Tool. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Statistics
The trim statistics per readset are merged at this step.
Bismark Aligner Step
In this step, reads are aligned using the Bismark Tool.
UMI BAM Tag Processing
This step uses fgbio tool to add read UMI tag to individual BAM files.
Sambamba Merge SAM Files
Sambamba is a powerful filtering tool used for extracting information from SAM/BAM files.
In this step, BAM readset files are merged into one file per sample. Merge is done using Picard Tool.
As input, this step takes the aligned and sorted BAM output files from previous step, UMI BAM Tag Processing, if available. Otherwise, it uses BAM files from the readset file as input.
Picard remove duplicates
This step removes duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be removed as a duplicate in the BAM file. Removing duplicates is done using Picard Tool.
Compute Metrics
Besides computing metrics, this step also generate coverage tracks per sample. Multiple metrics are computed at this stage: Number of raw reads, Number of filtered reads, Number of aligned reads, Number of duplicate reads, Median, mean and standard deviation of insert sizes of reads after alignment, percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads) whole genome or targeted percentage of bases covered at X reads (%_bases_above_50 means the % of exons bases which have at least 50 reads). A TDF (.tdf) coverage track is also generated at this step for easy visualization of coverage in the IGV browser.
Methylation Call
The script in this pipeline step reads in a bisulfite read alignment file produced by the Bismark bisulfite mapper and extracts the methylation information for individual cytosines. The methylation extractor outputs result files for cytosines in CpG, CHG and CHH context. It also outputs bedGraph, a coverage file from positional methylation data and cytosine methylation report.
Wiggle Tracks
This step generates wiggle tracks suitable for multiple browsers, to show coverage and methylation data. When using GRCh37 build of Human genome, to be compatible with the UCSC Genome Browser we only keep chromosomes 1-22, X, Y and MT, and we also rename them by prefixing “chr” to the chromosome name (e.g. “1” becomes “chr1”), and changing the mitochondrial chromosome from “MT” to “chrM”, and keeping the GRCh37 coordinates.
Methylation Profile
This step involves generation of a CpG methylation profile by combining both forward and reverse strand Cs.
It also generates all the methylation metrics:
CpG stats,
pUC19 CpG stats,
lambda conversion rate,
median CpG coverage,
GC bias
IHEC Sample Metrics Report
In this step, computed metrics which fit the IHEC standards, are retrieved. It also build a tsv report table for IHEC.
Bis SNP Processing
Identifying SNPs is important for accurate quantification of methylation levels and for identification of allele-specific epigenetic events such as imprinting. This step uses bisulfite SNP caller, Bis-SNP, for correctly identifying SNPs using the default high-stringency settings at an average 30× genomic coverage.
Filter SNP CpGs
This step involves SNP CpGs filtering. The goal here is to use a known variants db (Variant Call Format file provided via the ini file) to filter the CpG sites produced earlier during the step Methylation Profile.
Prepare for MethylKit Differential Analysis
This step enables comparison of methylation profile across samples. It prepares inputs that are required for the subsequent step of MethylKit Differential Analysis.
MethylKit Differential Analysis
This step involves running of MethylKit to get DMCs & DMRs for different designed comparisons.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
More information¶
For the latest implementation and usage details refer to WGS or Methylation Sequencing implementation README.md file.