RNA Sequencing (Light) Pipeline
This is a lightweight RNA Sequencing Expression analysis pipeline based on Kallisto technique. It is used for quick Quality Control (QC) in gene sequencing studies.
Introduction
The central computational problem in RNA-seq remains the efficient and accurate assignment of short sequencing reads to the transcripts they originated from and using this information to infer gene expressions. Conventionally, read assignment is carried out by aligning sequencing reads to a reference genome, such that relative gene expressions can be inferred by the alignments at annotated gene loci. These alignment-based methods are conceptually simple, but the read-alignment step can be time-consuming and computationally intensive.
Alignment-free RNA quantification tools have significantly increased the speed of RNA-seq analysis. The alignment-free pipelines are orders of magnitude faster than alignment-based pipelines, and they work by breaking sequencing reads into k-mers and then performing fast matches to pre-indexed transcript databases. To achieve fast transcript quantification without compromising quantification accuracy, different sophisticated algorithms were implemented in addition to k- mer counting, such as pseudo-alignments by Kallisto technique and quasi-mapping along with GC and sequence-bias corrections using Salmon.
RNA Sequencing Light is a lightweight pipeline that performs quick QC and removes a major computation bottleneck in RNA Sequence analysis. Kallisto is two orders of magnitude faster than previous approaches and achieves similar accuracy. Kallisto pseudo-aligns reads to a reference, producing a list of transcripts that are compatible with each read while avoiding the alignment of individual bases. In the latest release of GenPipes, calls to kallisto quant are now aggregated by sample instead of by the readset for better performance. For details, see ‘More Information’ section below.
Version
4.4.2
Usage
rnaseq_light.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [-d DESIGN]
[--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>]
[--genpipes_file GENPIPES_FILE] [-d DESIGN]
[-r READSETS] [-v]
Optional Arguments
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exita
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline".
Example Run
rnaseq_light.py -c $MUGQIC_PIPELINES_HOME/pipelines/rnaseq_light/rnaseq_light.base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini -r readset.rnaseq.txt -d design.rnaseq.txt -s 1-25 -g rnaseqCommands.sh
bash rnaseqCommands.sh
Tip
Replace beluga.ini file name in the command above with the appropriate clustername.ini file located in the $MUGQIC_PIPELINES_HOME/pipelines/common_ini folder, depending upon the cluster where you are executing the pipeline. For e.g., narval.ini, cedar.ini, graham.ini or narval.ini.
You can download the test dataset for this pipeline in the TestData Reference section
Pipeline Schema
Refer to the diagram below for the schema of RNA Sequencing (Light) Pipeline.
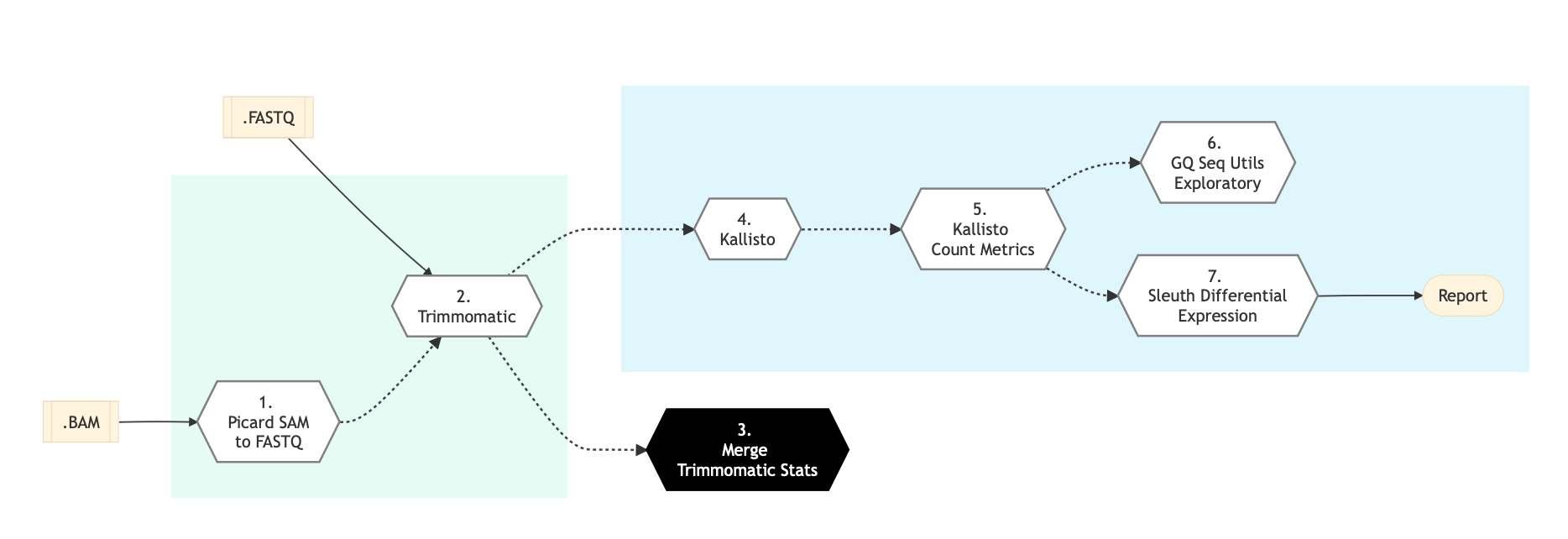
Pipeline Steps
| RNA Sequence Light Steps | |
|---|---|
| 1. | Picard SAM to FASTQ |
| 2. | Trimmomatic |
| 3. | Merge Trimmomatic Stats |
| 4. | Kallisto |
| 5. | Kallisto Count Matrix |
| 6. | GQ Seq Utils Exploratory |
| 7. | Sleuth Differential Expression |
Step Details
Picard SAM to FASTQ
Convert SAM/BAM files from the input readset file into FASTQ format if FASTQ files are not already specified in the readset file. Do nothing otherwise.
Trimmomatic
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Merge Trimmomatic Stats
The trim statistics per readset are merged at this step.
Kallisto
Run Kallisto on FastQ files for a fast estimate of abundance.
Kallisto Count Matrix
Use the output from Kallisto to create a transcript count matrix.
GQ Seq Utils Exploratory
Exploratory analysis using the gqSeqUtils R package adapted for RnaSeqLight.
Sleuth Differential Expression
Performs differential gene expression analysis using Sleuth. Analysis are performed both at a transcript and gene level, using two different tests: LRT and WT.
More Information
Kallisto, a new ultra-fast RNA Sequencing technique
Limitations of alignment-free tools in RNA sequencing quantification