Nanopore CoVSeQ Pipeline
GenPipes Nanopore CoVSeq pipeline is built using the Nanopore ARTIC-Nanopolish protocol. This protocol has been widely adopted by research groups worldwide to assist in epidemiological investigations. This protocol is mainly focused around the use of portable Oxford Nanopore MinION sequencer. However, other aspects of the protocol related to primer scheme and sample amplification can be generalized to other sequencing platforms.
Direct amplification of the virus using tiled, multiplexed primers approach has been proven to have high sensitivity. It enables researchers to work directly from clinical samples compared to metagenomic projects. It has been widely used to analyze viral genome data generated during outbreaks such as SARS-CoV-2 for information about relatedness to other viruses.
The GenPipes Nanopore CoVSeQ Sequencing Pipeline is based on nCoV-2019 novel coronavirus bioinformatics protocol (ARTIC V4.1) that takes the output from the sequencing protocol to consensus genome sequences. It includes basecalling, de-multiplexing, mapping, polishing and consensus generation.
Introduction
The Nanopore CoVSeQ pipeline is used to analyze long reads produced by the Oxford Nanopore Technologies (ONT) sequencers.
The SOP for Nanopore data is based on the ARTIC SARS-CoV2 protocol, Version 4 / 4.1 (V4.1), using nanopolish. This protocol is closely followed in GenPipes Nanopore sequencing pipeline with majority of changes related to technical adaptation of the protocol to be able to run in a High Performance Computing (HPC) environment. In such environments, Conda is not advisable.
Key steps in this pipeline include basecalling with Guppy, demultiplexing, read filtering and consensus sequencing. Basecalling with Guppy happens only if the `-t basecalling` option is selected.
If basecalling protocol option is selected through the -t command line option, the Nanopore CoVSeQ pipeline will do basecalling with Guppy (GPU) and demultiplexing. After basecalling, the pipeline performs de-hosting, for all the samples, followed by running the ARTIC-Nanopolish wrapper which performs alignment to the SARS-CoV2 reference (using minimap2), variant calling (using Nanopolish software). The Nanopolish software performs signal-level analysis of Oxford Nanopore sequencing data. After Nanopolish processing, the pipeline performs consensus generation through artic_mask and bcftools consensus steps. Lastly, custom scripts and ncov_tools are run to report on quality metrics for Nanopore CoVSeQ GenPipes Sequencing Pipeline.
Details of structure and contents of the Nanopore readset file are available here.
Version
4.5.0
For the latest implementation and usage details refer to Nanopore Sequencing implementation README file file.
Usage
nanopore_covseq.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug, info, warning, error, critical}] [--sanity-check]
[-t {default (default), basecalling}]
[--genpipes_file GENPIPES_FILE]
[--container {wrapper, singularity} {<CONTAINER PATH>, <CONTAINER NAME>}]
[-v]
Optional Arguments
-t {default, basecalling}, --type {default, basecalling}
Nanopore Covseq analysis type (default=default)
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
Note: Do not use -g option with --clean, use '>' to redirect
the output of the --clean command option
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exit
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline"
Note: Do not use -g option with -clean. Use '>' to redirect
the output to a file when using -clean option.
-r READSETS, --readsets READSETS
readset file
Example Run
Use the following commands to execute Nanopore sequencing pipeline:
nanopore_covseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/nanopore/nanopore.base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini $MUGQIC_PIPELINES_HOME/pipelines/nanopore_covseq/ARTIC_v4.1.ini -g nanopore_covseq_commands_mugqic.sh
bash nanopore_covseq_commands.sh
Tip
Replace beluga.ini file name in the command above with the appropriate clustername.ini file located in the $MUGQIC_PIPELINES_HOME/pipelines/common_ini folder, depending upon the cluster where you are executing the pipeline. For e.g., narval.ini, cedar.ini, graham.ini or narval.ini.
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
Warning
ARTIC v4 vs v4.1 selection
The Nanopore CoVSeQ pipeline uses ARTIC v4 amplicon scheme as a default. If ARTIC v4.1 is required, use the appropriate .ini file. For all other amplicon schemes, add the appropriate primer and amplicon bed files and use a custom .ini for processing.
Note
Nanopore Readset Format
Use the following readset file format for Nanopore, Nanopore CoV-Seq pipelines:
Sample: must contain letters A-Z, numbers 0-9, hyphens (-) or underscores (_) only; mandatory;
Readset: a unique readset name with the same allowed characters as above; mandatory;
Run: a unique ONT run name, usually has a structure similar to PAE0000_a1b2c3d;
Flowcell: code of the type of Flowcell used, for example: the code for PromethION Flow Cell (R9.4) is FLO-PRO002;
Library: code of the type of library preparation kit used, for example: the code for the Ligation Sequencing Kit is SQK-LSK109;
Summary: path to the sequencing_summary.txt file outputted by the ONT basecaller; mandatory;
FASTQ: mandatory, path to the fastq_pass directory, that is usually created by the basecaller
FAST5: path to the directory containing the raw fast5 files, before basecalling
Example:
Sample |
Readset |
Run |
Flowcell |
Library |
Summary |
FASTQ |
FAST5 |
sampleA |
readset1 |
PAE00001_abcd123 |
FLO-PRO002 |
SQK-LSK109 |
path/to/readset1_sequencing_summary.txt |
path/to/readset1/fastq_pass |
path/to/readset1/fast5_pass |
sampleA |
readset2 |
PAE00002_abcd456 |
FLO-PRO002 |
SQK-LSK109 |
path/to/readset2_sequencing_summary.txt |
path/to/readset2/fastq_pass |
path/to/readset2/fast5_pass |
sampleA |
readset3 |
PAE00003_abcd789 |
FLO-PRO002 |
SQK-LSK109 |
path/to/readset3_sequencing_summary.txt |
path/to/readset3/fastq_pass |
path/to/readset3/fast5_pass |
sampleA |
readset4 |
PAE00004_abcd246 |
FLO-PRO002 |
SQK-LSK109 |
path/to/readset4_sequencing_summary.txt |
path/to/readset4/fastq_pass |
path/to/readset4/fast5_pass |
You can download the test dataset for this pipeline here.
Pipeline Schema
Figure below shows the schema of the Nanopore CoVSeQ ARTIC SARS-CoV2 sequencing protocol. You can also refer to the latest pipeline implementation
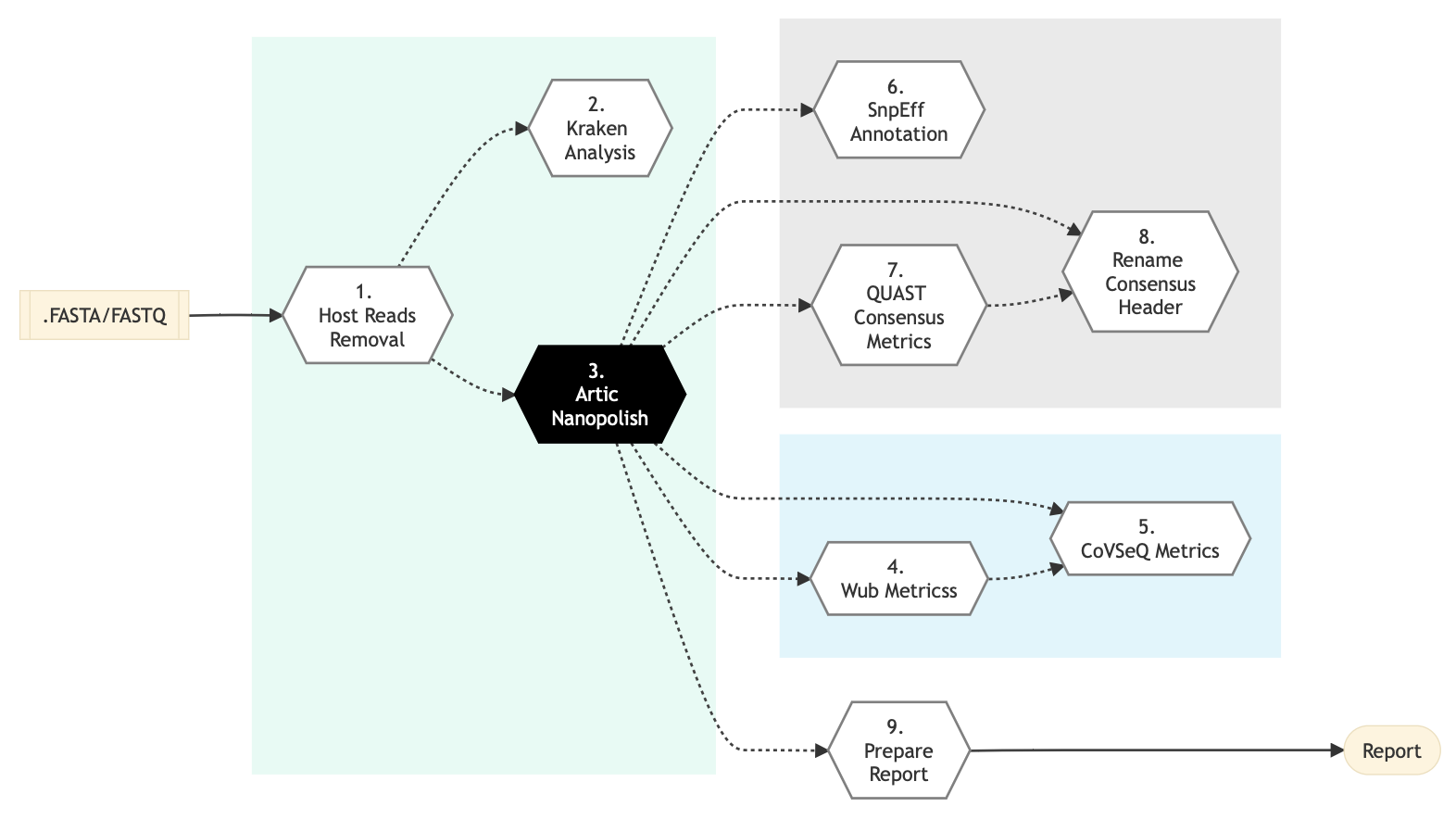
Figure: Schema of Nanopore CoVSeQ (Default) Sequencing protocol
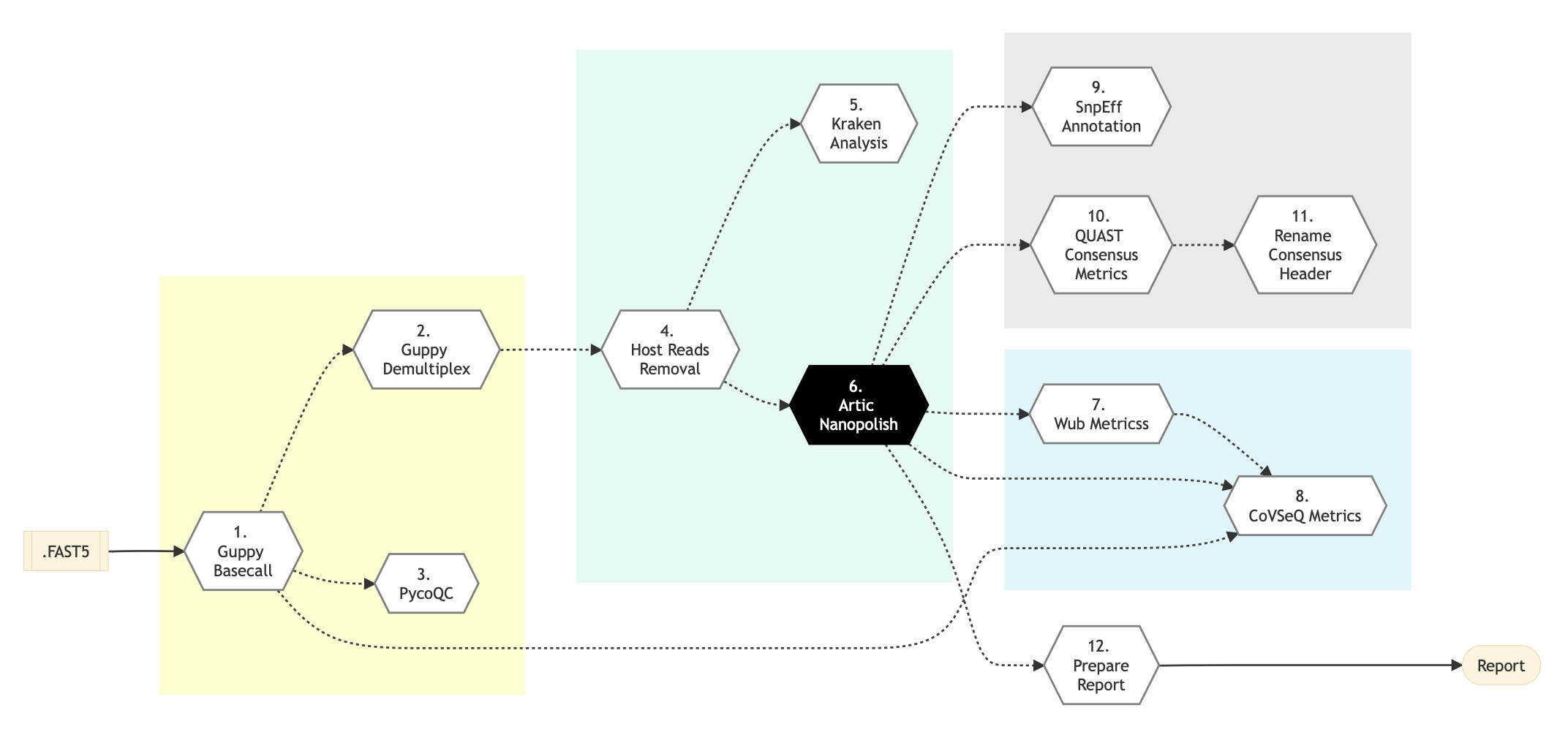
Figure: Schema of Nanopore CoVSeQ (Basecalling) Sequencing protocol
Pipeline Steps
The table below shows various steps that constitute the Nanopore CoVSeQ ARTIC-v4.1 analysis pipeline.
Default Nanopore CoVSeQ | Basecalling Nanopore CoVSeQ |
||
|---|---|---|
Step Details
Following are the various steps that are part of GenPipes Nanopore CoVSeQ genomic analysis pipeline:
Guppy Basecall
This step uses the Oxford Nanopore basecaller, Guppy to basecall raw FAST5 files and produce FASTQ files. Basecalling model dna_r9.4.1_450bps_hac.cfg is used by default.
Guppy Demultiplex
This step uses he Oxford Nanopore basecaller Guppy to demultiplex FASTQ files based on their barcode. Barcode arrangement barcode_arrs_nb96.cfg is used by default.
Note
In the Guppy Demultiplex call, the following parameter, `--require_barcodes_both_ends`, is set by default.
pycoQC
In this step, pycoQC Software is used produce an interactive quality report based on the summary file and alignment outputs. PycoQC relies on the sequencing_summary.txt file generated by Guppy. If needed, it can also generate a summary file from basecalled FAST5 files. PycoQC computes metrics and generates interactive QC plots for Oxford Nanopore technologies sequencing data.
Host Reads Removal
This step uses a mapping approach with a hybrid GRCh38 + SARS-CoV2 genome. The reads that map to the Human Genome are removed from the analysis. A “de-hosted” FASTQ is produced.
Kraken Analysis
Kraken is a taxonomic sequence classifier that assigns taxonomic labels to short DNA reads. It does this by examining the k-mers within a read and querying a database with those k-mers.
Additionally, Kraken2 is used to produce a report on the raw data, which can be used to detect additional host contamination.
ARTIC Nanopolish
The ARTIC Nanopolish pipeline is used to produce consensus sequences and VCFs. Since Nanopolish is used, this step requires both FAST5 and FASTQ files.
Wub Metrics
Wub Package is used to calculate alignment metrics in this pipeline step.
CoVSeQ Metrics
Using all previous metrics calculated so far, a table is produced with a summary of all metrics for each individual sample.
SnpEff Annotate
The VCF produced by ARTIC Nanopolish step is annotated using SnpEff.
Quast Consensus Metrics
Consensus metrics are calculated using the tool QUAST.
Rename Consensus Header
A final consensus sequence is produced, with the appropriate header and naming convention based on genome completeness.
Prepare Report
Using ncov-tools package and additional R scripts, final reports are produced for all samples in the run, including basic QC plots as well as a preliminary lineage assignment through ncov-tools package.
More information
For the latest implementation and usage details refer to Nanopore CoVSeq Pipeline implementation README.md.
Phylogenetic Analysis of nCoV-2019 genome using publicly shared genome sequences with datasets from NCBI or GISAID.
Tiling Amplicon sequencing and downstream bioinformatics analysis