EpiQC Pipeline
The epiQC is a quality control pipeline for signal files (BigWig) generated during ChIP-Sequencing Pipeline execution. The epiQC pipeline performs a series of calculations on these BigWig files to assess the quality of ChIP-Seq data. Low quality bases can bias various genomic analysis processes such as SNP and SV calling. The epiQC Pipeline helps in determining the quality of inputs.
As part of epiQC pipeline, four different metrics are computed from a single BigWig file.
BigWigInfo tool prints out information about a BigWig file. This tool is used to perform the initial quality check on signal tracks.
ChromImpute processing step imputes signal tracks for the given chromosome (currently only chr1 is supported, but it is sufficient to accurately detect issues in signal files).
Signal to Noise step of the epiQC pipeline processes the signal to noise measurement by calculating the proportion of signal in top bins.
This is followed by heatmap creation from the correlation matrix obtained via the epiGenomic Efficient Correlator (epiGeEC) tool by comparing only the user samples.
Note
At present, comparing large reference database with user samples is not supported.
After the completion of computing metrics, the epiQC pipeline executes four consecutive report steps to create the epiQC Final Report of the pipeline with quality control labels.
This brand new epiQC pipeline can be used for pre-validation of samples, in order to assess the usability of a dataset in any given study, even in the absence of the original raw reads files. This presents a huge advantage, for instance, in the case of human epigenomic datasets available in the IHEC Datasets, as signal tracks are made publicly available, while raw data files are stored in controlled access repositories.
You can test this pipeline with ChIP-Seq samples from the IHEC Portal.
For more information on epiQC pipeline implementation and design, refer to the README.md file.
Introduction
GenPipes introduces epiQC as a brand new pipeline with the sole focus of quality control for BigWig signal files. These BigWig signal files are generated as part of ChIP-Sequencing Pipeline. It is important to determine the quality of bases before using them for subsequent analysis as low quality bases can bias the downstream analysis such as SNP and SV calling.
The following sections describe pipeline usage and various steps in the epiQC pipeline.
Version
4.5.0
For the latest implementation and usage details refer to epiQC Pipeline implementation README file.
Usage
epiqc.py [-h] [--help] [-c CONFIG [CONFIG ...]] [-s STEPS]
[-o OUTPUT_DIR] [-j {pbs,batch,daemon,slurm}] [-f]
[--no-json] [--report] [--clean]
[-l {debug,info,warning,error,critical}] [--sanity-check]
[--container {wrapper, singularity} <IMAGE PATH>]
[-r READSETS] [-v]
[--genpipes_file GENPIPES_FILE]
Optional Arguments
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...], --config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm}, --job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
--no-json do not create JSON file per analysed sample to track
the analysis status (default: false i.e. JSON file
will be created)
--report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
Note: Do not use -g option with --clean, use '>' to redirect
the output of the --clean command option
-l {debug,info,warning,error,critical}, --log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
-v, --version show the version information and exit
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline"
Note: Do not use -g option with -clean. Use '>' to redirect
the output to a file when using -clean option.
Example Run
Use the following commands to execute epiQC pipeline:
epiqc.py -c $MUGQIC_PIPELINES_HOME/pipelines/epiqc/epiqc.base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini -g epiqcCommands_mugqic.sh
bash epiqcCommands_mugqic.sh
Tip
Replace beluga.ini file name in the command above with the appropriate clustername.ini file located in the $MUGQIC_PIPELINES_HOME/pipelines/common_ini folder, depending upon the cluster where you are executing the pipeline. For e.g., narval.ini, cedar.ini, graham.ini or narval.ini.
Warning
While issuing the pipeline run command, use `-g GENPIPES_FILE` option (see example above) instead of using the ` > GENPIPES_FILE` option supported by GenPipes so far, as shown below:
[genpipes_seq_pipeline].py -t mugqic -c $MUGQIC_PIPELINES_HOME/pipelines/[genpipes_seq_pipeline]/[genpipes_seq_pipeline].base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini -r readset.[genpipes_seq_pipeline].txt -s 1-6 > [genpipes_seq_pipeline]_commands_mugqic.sh
bash [genpipes_seq_pipeline]_commands_mugqic.sh
` > scriptfile` should be considered deprecated and `-g scriptfile` option is recommended instead.
Please note that redirecting commands to a script `> genpipe_script.sh` is still supported for now. But going forward, this mechanism might be dropped in a future GenPipes release.
You can download the test dataset for this pipeline here.
Pipeline Schema
Figure below shows the schema of the epiQC pipeline.
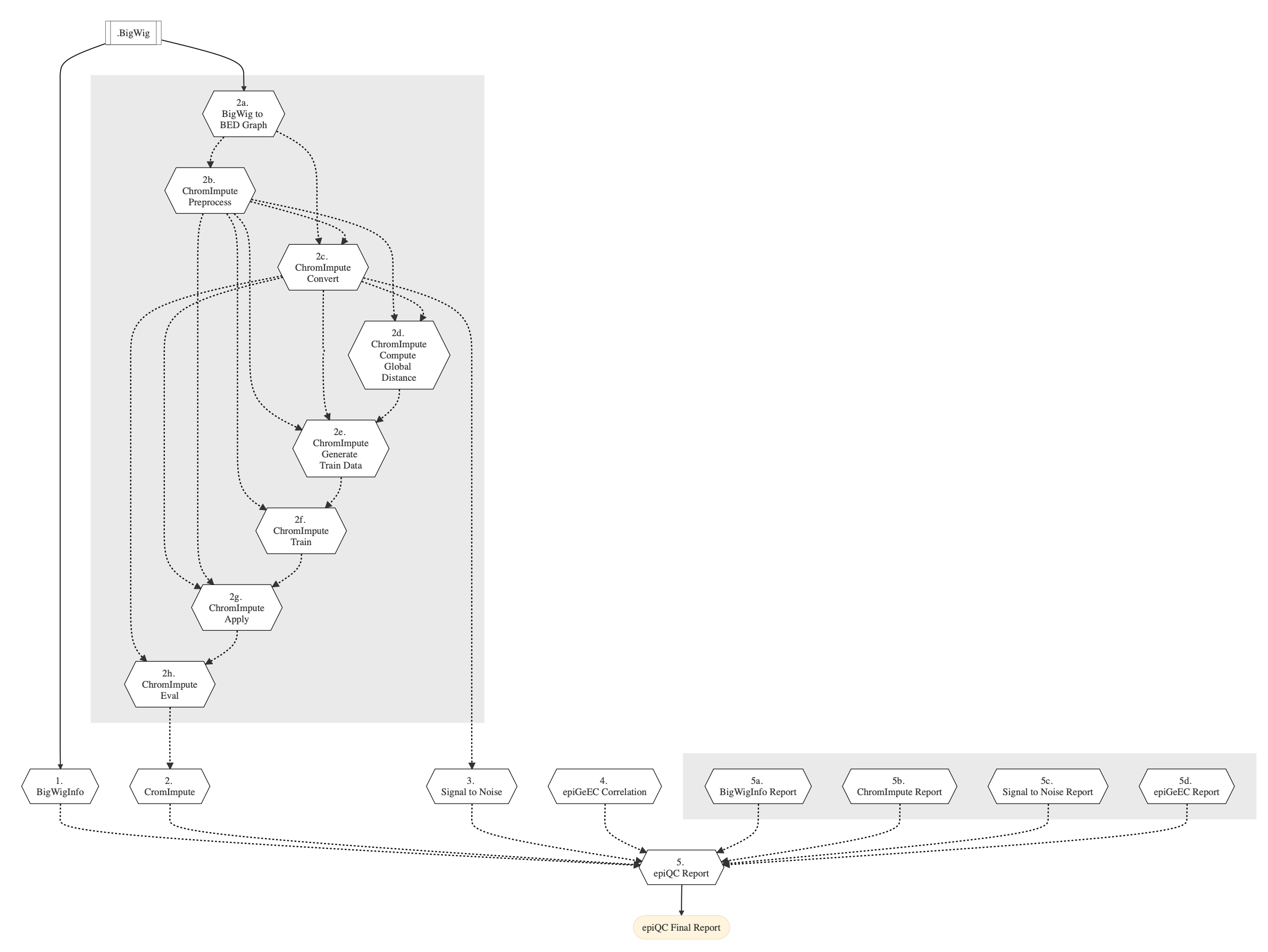
Figure: Schema of epiQC pipeline
Pipeline Steps
The table below shows various steps that constitute the epiQC pipeline.
epiQC Pipeline Steps |
||
|---|---|---|
Step Details
Following are the various steps that are part of GenPipes epiQC pipeline:
BigWigInfo Processing
This step takes BigWig file as input and runs the BigWigInfo Tool to inspect signal tracks and identify obvious issues that may impact the quality of the ChIP-Seq data. BigWigInfo is capable of identifying various obvious issues such as missing chromosomes and insufficient track coverage. These are usually symptoms of improperly generated tracks.
If the user has specified BigWig files in the readset file under BIGIWIG column, they will be utilized by the tool. Otherwise, the user is required to process files using ChIp-Seq pipeline to generate these BigWig files that are required as inputs for epiQC Pipeline. In this case, the paths for BigWig files are reconstructed based on the ChIP-Seq readset file and will be used subsequently.
Warning
Location of readset file
The readset file should be in the same folder as the ChIP-Seq output.
ChromImpute Processing
ChromImpute is a Java software for large-scale systematic epigenome imputation. ChromImpute takes an existing compendium of signal tracks (BedGraph Files) and uses it for:
Predicting signal tracks for mark-sample combinations that are not experimentally mapped.
Generates a potentially more robust version of data sets that have been mapped experimentally.
ChromImpute bases its predictions on features from signal tracks of other marks that have been mapped in the target sample and the target mark in other samples with these features combined using an ensemble of regression trees. For better results, usage of multiple histone marks from one sample is recommended.
Note
Support for GRCh38, GRCh37
The current epiQC Pipeline implementation supports only those signal tracks that are mapped to GRCh38 during ChromImpute analysis. GRCh37 will be added in the future. For better results, usage of multiple histone marks from one sample is recommended.
ChromImpute processing step comprises of several sub-steps that are invoked internally in the pipeline.
Refer to the section below for the sub-step details:
BigWig to BED Graph
In this sub-step of ChromImpute Processing, BigWig file is the input to the BigWigtoBEDGraph Tool for converting BigWig files to BedGraph Files. These BedGraph files are used in other ChromImpute steps subsequently.
ChromImpute Preprocessing
This sub-step of ChromImpute Processing is performed to create ChromImpute directories, chromosome sizes file, inputinfo file with IHEC and user samples. Finally, this step also links the converted IHEC BedGraph Files to user directory. In order to run the ChromImpute, it is required that the inputinfo and chromsizes file are located in the imputation directory.
Note
The chromsizes and inputinfo files are created dynamically when the user runs the epiQC pipeline. It is not necessary to submit jobs to create them.
Although the current implementation supports only GRCh38 chr1 for ChromImpute analysis, it is sufficient to accurately evaluate the signal quality. Chromosome name can be specified in the ChromImpute Preprocessing section of the epiQC pipeline .ini file.
ChromImpute Convert
The ChromImpute convert sub-step is performed to convert each unique mark and sample combination signal tracks (BedGraph Files) in the user dataset into binned (25 bp) signal resolution tracks. Since the stored, converted files are binned using 25bps, changing the resolution value specified in the epiQC pipeline .ini file is not recommended for ChromImpute analysis.
Note
If you encounter an ‘index out of bound exception’, check whether your reference genome version of the BedGraph file is similar to chromosome_sizes_file inside the imputation folder.
The ChromImpute convert sub-step takes as input files:
BigWig files from BigWig to BED Graph sub-step earlier.
Chromosome sizes file
inputinfo file
ChromImpute Compute Global Distance
This sub-step is used to compute the global distance based on correlation for each mark in each sample with the same mark in all other samples in inputinfo file. It creates a file for each mark, in each sample containing a ranked list of the globally nearest samples.
This sub-step takes as input the following files:
Converted signal tracks from ChromImpute Convert sub-step
Converted and linked signal tracks from IHEC data portal
Chromosome sizes file
inputinfo file
ChromImpute Generate Train Data
This step is performed to generate a set of training data instances taking directory of converted data and global distances.
This step takes as input the following files:
Converted signal tracks from ChromImpute Convert.
Converted and linked signal tracks from IHEC data portal
Chromosome sizes file
inputinfo file
global distance files from ChromImpute Compute Global Distance sub-step.
ChromImpute Train
This step is used to train regression trees based on the feature data produced by ChromImpute Generate Train Data.
This step takes as input files:
Predictor data produced by ChromImpute Generate Train Data sub-step.
Chromosome sizes file
inputinfo file
ChromImpute Apply
This step is used to apply the predictors generated in the Train command to generate the imputed data. A job is created for each sample and mark given in the dataset.
This sub-step takes as input files:
Converted signal tracks from ChromImpute Convert
Converted and linked signal tracks from IHEC data portal
Chromosome sizes file
inputinfo file
global distance files from ChromImpute Compute Global Distance
Predictor data produced by ChromImpute Generate Train Data
ChromImpute Evaluation
The final sub-step of ChromImpute Processing involves comparing the agreement between an observed and imputed data set. A job is created for every sample-mark given in the dataset. percent1 percent2 Gives lower and upper percentages to use in evaluation. Default values for percent1 is 1% and percent2 is 5%
This step takes as input files:
Converted signal tracks from ChromImpute Convert
Imputed signal tracks from ChromImpute Apply
Signal To Noise
Binned signal resolution tracks from ChromImpute Convert step is used to determine the percentage of the whole file signal that was located in the top percent1 and percent2 of the bins. The default percentages are 5% and 10% and the resulted output is a tsv file.
This step takes converted signal tracks from ChromImpute Convert as input file.
epiGeEC Processing
This step is performed to run the EpiGeEC pipeline. EpiGeEC pipeline consists of three sub-steps:
Bigwig files are first converted to the hdf5 format.
Next, it filters or selects the provided regions as a BED file (include or exclude) [optional]. The user can specify the options and the BED file path in the epiQC pipeline .ini file. Otherwise, this step will be skipped
Finally, the correlation matrix is computed.
epiQC Report
epiQC report comprises of five sub-steps including four independent steps to generate individual report files for each metric that is computed and one step to combine all of them to generate a final epiQC report file. A user can independently run each sub step but in order to generate final report, there is a dependency on the reports generated from BigWigInfo Processing, ChromImpute Processing and EpiGeEC Processing steps.
This final report is a TSV file with the decision of each signal track [i.e whether the sample has passed based on all the metrics involved in various reports or are there any alerts that the user need to be concerned about.]
BigWigInfo Report
This step is performed to generate report on bigwiginfo result
ChromImpute Report
This step is performed to generate a report comparing ChromImpute imputed signal track and input signal track (in bedgraph format).
Signal to Noise Report
This step is performed to generate report on signal_to_noise result
epiGeEC Report
This step is performed to generate a heatmap from EpiGeEC results
epiQC Final Report
Once all metrics have been obtained, they are gathered in a TSV formatted report and compared to predetermined thresholds. A column holding epiQC’s verdict for a signal track is also included, using these thresholds evaluations.
The following metrics threshold were used:
Note
An Alert will be generated even if there is an issue with one of the several quality metrics.
High Level Alert:
Chromosome count is under 23
Medium Level Alert:
Whole genome bases covered under 25,000,000
Signal in top 10% bins below 30%
ChromImpute OBSERVED_1.0_IMPUTE_5.0 below 30%
Low Level Alert:
Whole genome bases covered under 75,000,000
Signal in top 5% bins below 20%
ChromImpute BOTH_1.0 below 20%
Refer to the figure below for insights related to probability of error and threshold levels:
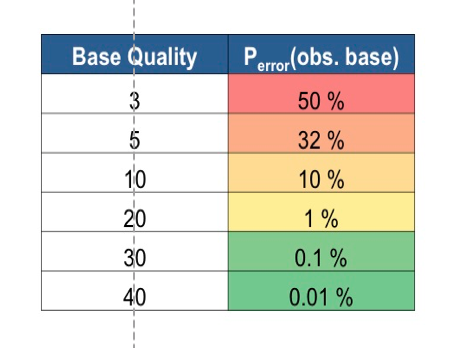
Figure: Base Quality Threshold and error probability
Note
In the current implementation of the epiQC pipeline, the epiGeEC metric is not used for the final quality decision.
More information
For the latest implementation and usage details refer to epiQC Pipeline implementation README.md.
epiGeEC Tool.
About ChromImpute.