New Users
To create a new CCDB account, what should I fill in the form field: ‘position’?
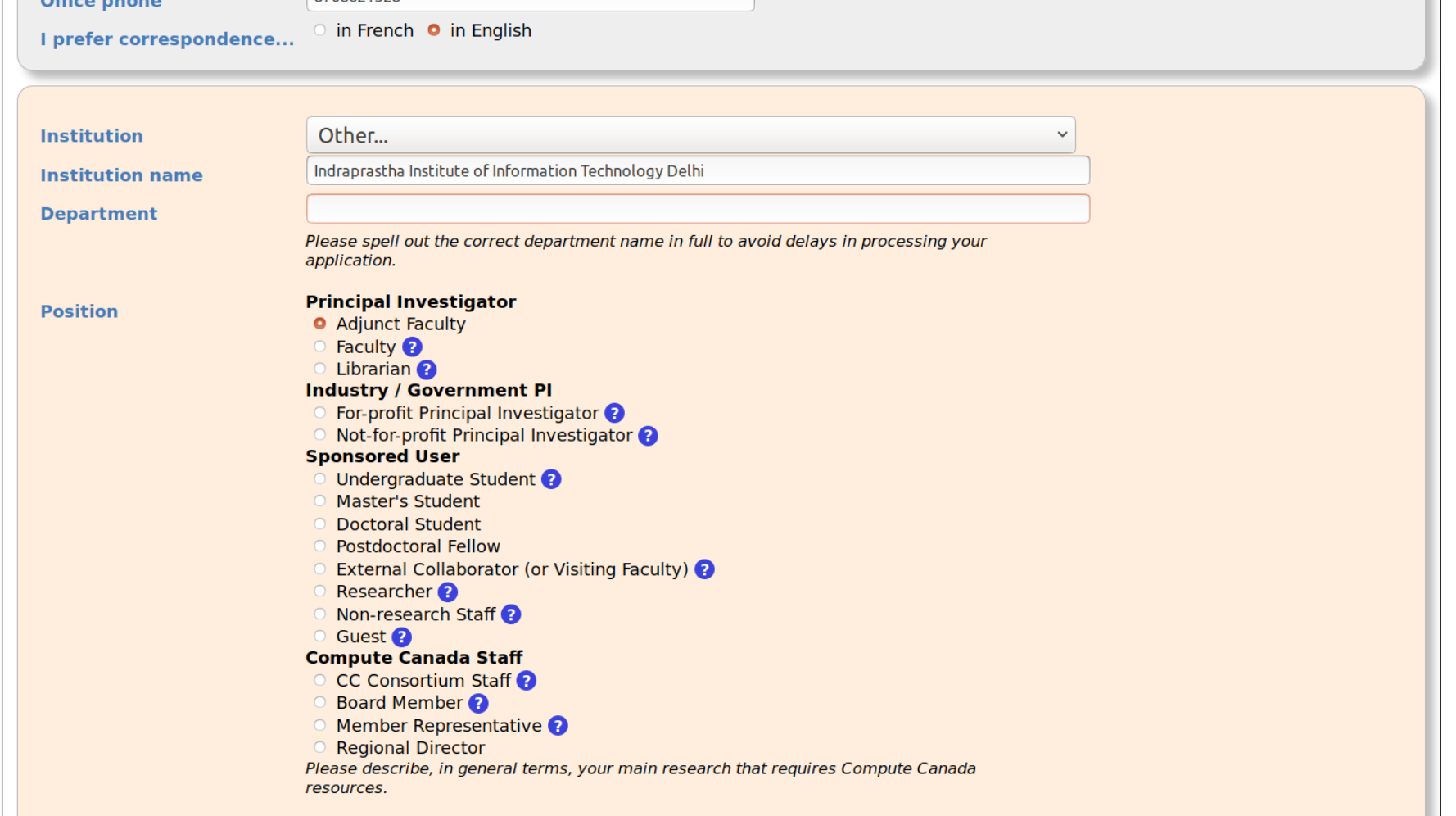
Response
Choose an appropriate option in the form, for example:
external collaborator
For the CCRI field, use the your sponsor’s Compute Canada Role Identifier (CCRI) as input. The CCRI has a structure similar to this: abc-123-01.
What email ID should I use if I am an external collaborator?
Response
GMail or your work ID should be fine as long as you provide the name of your institution (or college in case of students).
My account is activated. How do I learn more about Compute Canada Servers and resources available?
Response
My account is activated but I cannot login into beluga-computecanada or any other node - Cedar, Niagra? What is wrong?
Response
Check out the current CCDB server status here. Many a times, not being able to log in might just be due to system unavailability.
Please note that if you try to log in 3 or more times consecutively with a wrong password, your account gets deactivated and your IP address might get blacklisted. You would need to write to Compute Canada Support to get that reversed.
What is the best place to report GenPipes bugs?
Response
The preferred way to report GenPipes bugs is on our Bitbucket issue page : https://bitbucket.org/mugqic/genpipes/issues.
You can always reach us by email at mailto:pipelines@computationalgenomics.ca or use Google Group for GenPipes for any requests.
For more details, visit GenPipes Support and GenPipes Channels page in this documentation.
I was trying to use GenPipes deployed in a Docker Container. What <tag> value should I use?
Response
You can use the “latest” tag or one of the tags listed at GenPipes Docker Hub:. If you omit the <tag> Docker will use “latest” by default.
How do I run GenPipes locally in my infrastructure using a containerized setup? Is a scheduler setup necessary?
GenPipes pipelines use scheduler’s calls (qsub, sbatch) for submitting genomic analysis compute jobs. If you plan to use GenPipes locally using your infrastructure, inside a container, you need to run the GenPipes pipeline python scripts using the “batch mode” option. For local containerized versions of GenPipes, this is the preferred way of running the pipelines, if you don’t have access to a scheduler locally such as SLURM or PBS.
This is how you can run GenPipes pipelines such as DNA Sequencing Pipeline, refer to the command below:
dnaseq.py -c dnaseq.base.ini dnaseq.batch.ini -j batch -r your-readsets.tsv -d your-design.tsv -s 1-34 -t mugqic > run-in-container-dnaseq-script.sh
bash run-in-container-dnaseq-script.sh
Please note, there is a disadvantage to running GenPipes Pipelines without a scheduler. In the batch mode, which is configured using the “-j batch” option, all the jobs would run as a batch, one after another, on a single node. If your server is powerful enough, this might be your preferable option. Otherwise, if you would like to take advantage of GenPipes’ job scheduling capabilities, you need to install a job scheduler locally in your infrastructure so that GenPipes can work effectively. We recommend SLURM scheduler for GenPipes.
GenPipes 3.4 RNA Sequencing Pipeline moved from using Trimmomatic to Skewer. Why? What does this change mean for GenPipes users?
In addition to why, there are additional queries that we received. So we will respond to all of these together:
In RNA-seq skewer, a. What does the untrimmed read pairs available after processing refer to? b. If a large proportion of the reads are untrimmed does this mean the adapter sequence is wrong and how to troubleshoot this issue?
Why the switch?
The switch from trimmomatic to skewer was based on benchmarking. Skewer had improved F1 score across numerous truth sets over trimmomatic.
Untrimmed Reads and Troubleshooting
Untrimmed read pairs refers to read pairs which did not require quality 3’ trimming i.e. the quality was above 25 phred score or were above the size selection criteria of 50 bp after trimming and/or adapter removal.
Typically when the fastqc are generated after sequencing the adapters are removed, but in some cases the adapter remains. You can use fastqc on the raw reads to visualize the proportion of these. Also if you are unsure check that the adapters you are using are inline with sequencer and libraries you are using.