GenPipes Developers
How do I modify parameters/ options for specific tools or change computational resources that require a job to run?
GenPipes is fairly flexible in terms of the tooling used. Users can specify requisite parameters to use specific tools such as schedulers, python language version etc.
To change computational resources or use specific tools, you would need to create a custom config or ini file. Add the corresponding section you need to modify and change the parameters.
For example, if you want to change parameters of the macs2_callpeak step in the ChIPSeq pipeline, first create a new text file called custom.ini or you can use any name you prefer. Copy what is already in the chipseq.base.ini for this step and paste it in your custom.ini file.
[macs2_callpeak]
# Mandatory for module_macs2=mugqic/MACS2/2.2.7.1
module_python=mugqic/python/3.7.3
# The arbitrary shift in bp (Default for ATAC-Seq 75 ; Default for ChIP-Seq Narrow mark 0 ; Default for ChIP-Seq Broad mark 0)
shift=
# The arbitrary extension size in bp (Default for ATAC-Seq 150 ; Default for ChIP-Seq Narrow mark 200 ; Default for ChIP-Seq Broad mark 200)
extsize=
# Pvalue cutoff for peak detection (Default for ATAC-Seq 0.01 ; Default for ChIP-Seq Narrow mark `not set` ; Default for ChIP-Seq Broad mark `not set`)
pvalue=
other_options =
cluster_mem = 32G
cluster_cpu = 2
cluster_walltime = 12:00:0
The content above are the default settings, you can change them as shown below. For example, if you want to filter MACS2 peaks by FDR 0.01, then you should add -q 0.01 to the other_option section. Also, you can change the memory and cluster walltime to 64GB and 24 hours respectively. You can remove all the parameters you don’t want to change. The final file will look as shown below.
[macs2_callpeak]
other_options =-q 0.01
cluster_mem = 64G
cluster_walltime = 24:00:00
Now save the updated custom.ini file and mention it on the command line after the cluster ini file when you run GenPipes. The order of the custom ini file is important to override default parameters. Therefore, please make sure to add your custom.ini file at the end of all the other ini files.
hipseq.py -c $MUGQIC_PIPELINES_HOME/pipelines/chipseq/chipseq.base.ini $MUGQIC_PIPELINES_HOME/pipelines/common_ini/beluga.ini custom.ini -r readset.chipseq.txt -d design.chipseq.txt -s 1-20 -g chipseqScript.txt
Pytest command on CCDB server results in command not found error. Pytest install fails.
A new developer trying to setup and run GenPipes tests found the following issues:
I am trying to run some python test cases using "pytest" on beluga cluster.
I am running into "pytest command not found " error.
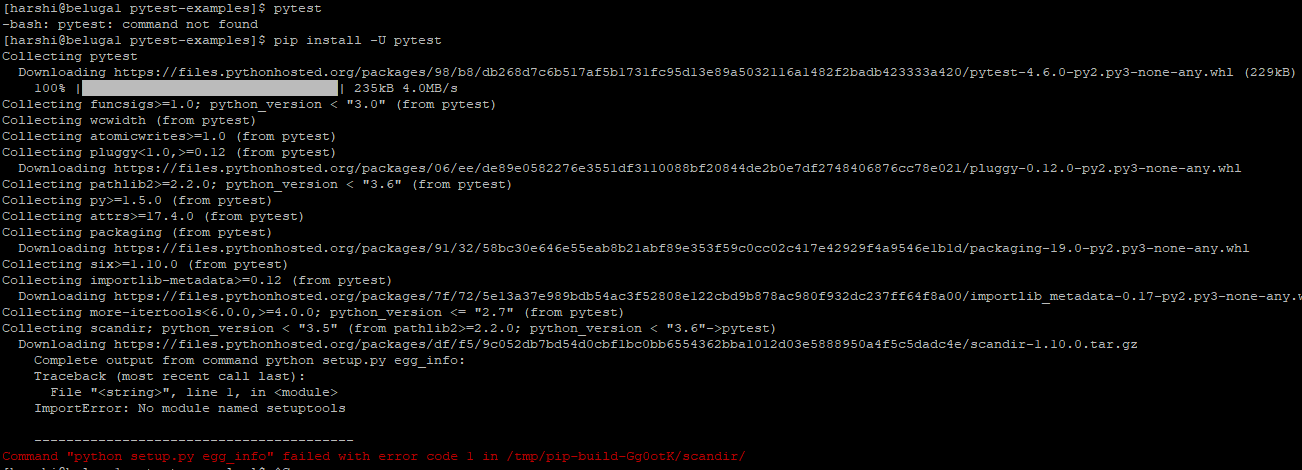
I googled that error and found that it might be due to older version of setup tools.
I tried to upgrade it and I'm seeing this error now.
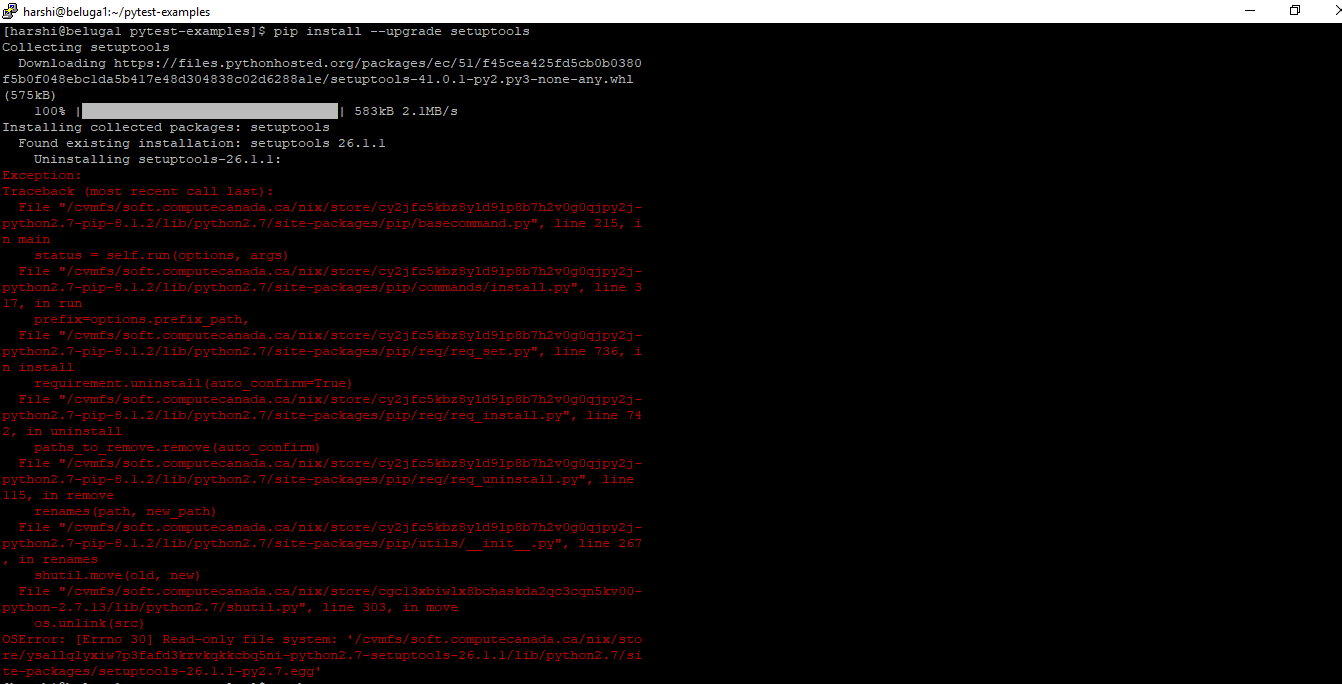
Response
Once your account is activated, you can login in CCDB servers such as Beluga, Cedar, Niagra. However, these are National Systems on a shared grid and users don’t have permission to install or upgrade the software there.
For more information on what software is installed on Compute Canada infrastructure, refer to https://docs.computecanada.ca/wiki/Available_software.
What kind of further analysis be done using HiC sequencing Pipeline?
Response
GenPipes Real Life Applications section of the documentation states that the Hi-C pipeline’s further analyses may integrate ChIP-seq/RNA-seq data. Refer to the Bait Intersect and Capture Intersect steps of the pipeline. These steps help to integrate ChIP-seq / RNA-seq data. You can see which of your regions have ChIP-seq or RNA-seq signal.