RNA Sequencing Pipeline
Version 6.1.0
Command
user@machine:~$ genpipes rnaseq [options] [--genpipes_file GENPIPES_FILE.sh]
user@machine:~$ bash GENPIPES_FILE.sh
Options
-t {stringtie, variants, cancer},
--type {stringtie, variants, cancer}
Type of RNA-seq assembly method (default=stringtie)
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...],
--config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm},
--job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
Take the mem input in the ini file and force to have a
minimum of mem_per_cpu by correcting the number of cpu
(default: None)
--force_mem_per_cpu FORCE_MEM_PER_CPU
Take the mem input in the ini file and force to have a
minimum of mem_per_cpu by correcting the number of cpu
(default: None)
--json-pt create JSON file for project_tracking database
ingestion (default: false i.e. JSON file will NOT be
created)
- -report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
Note: Do not use -g option with --clean, use '>' to redirect
the output of the --clean command option
-l {debug,info,warning,error,critical},
--log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
--wrap [WRAP] path to the GenPipes cvmfs wrapper script (default:
genpipes/ressources/container/bin/container_wrapper.sh,
a convenience option for using GenPipes in a container)
-v, --version show the version information and exit
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline"
Important
Do not use -g option with -clean.
Use ‘>’ to redirect the output of the genpipes command to a file when using -clean option.
Example
user@machine:~$ genpipes rnaseq -c $GENPIPES_INIS/rnaseq/rnaseq.base.ini \ $GENPIPES_INIS/common_ini/rorqual.ini \ -r readset.rnaseq.txt \ -d design.rnaseq.txt \ -s 1-21 \ -g rnaseqCommands.sh user@machine:~$ bash rnaseqCommands.sh
Tip
Depending upon the cluster where you are executing the pipeline, substitute the file name rorqual.ini in the command with the appropriate <DRAC server cluster name>.ini file located in the $GENPIPES_INIS/common_ini folder.
For e.g., rorqual.ini, fir.ini, or narval.ini.
Caution
It is recommended that you use the -g GENPIPES_CMD.sh option instead of redirecting the output of the pipeline command to a file via > GENPIPES_CMD.sh.
user@machine:~$ genpipes [pipeline] [options] -g genpipes_cmd.sh
user@machine:~$ bash genpipes_cmd.sh
user@machine:~$ genpipes [pipeline] [options] > genpipes_cmd.sh
user@machine:~$ bash genpies_cmd.sh
The > scriptfile method is supported but will be deprecated in a future GenPipes release.
You can download the test dataset for this pipeline. For more details, you can refer to GenPipes RNA Sequencing Workshop 2018 presentation.
StringTie
The following figure shows the schema of the RNA sequencing protocol (stringtie).
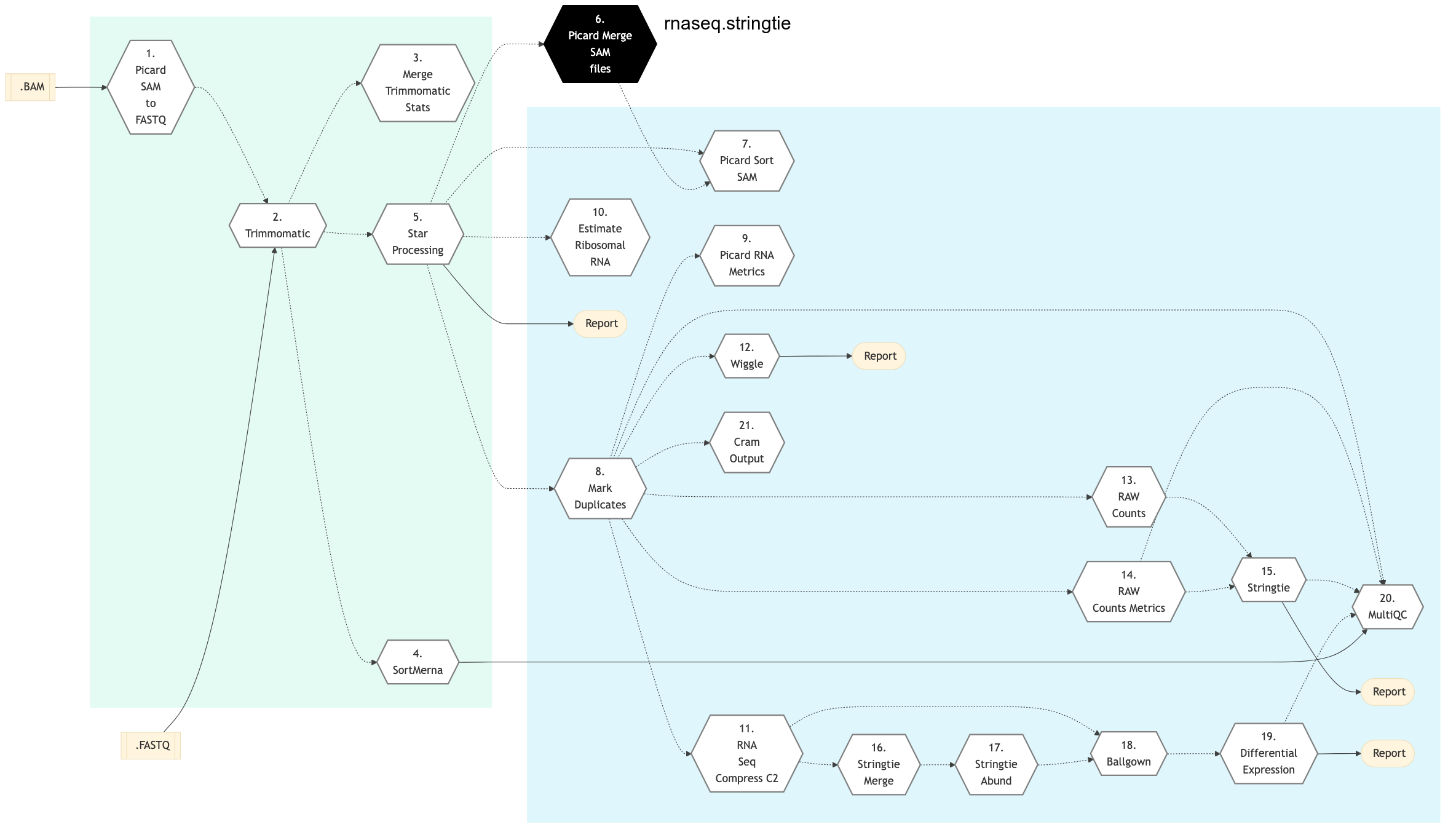
Figure: Schema of RNA Sequencing pipeline (StringTie)
Variants
The following figure shows the schema of the RNA sequencing protocol (variants).
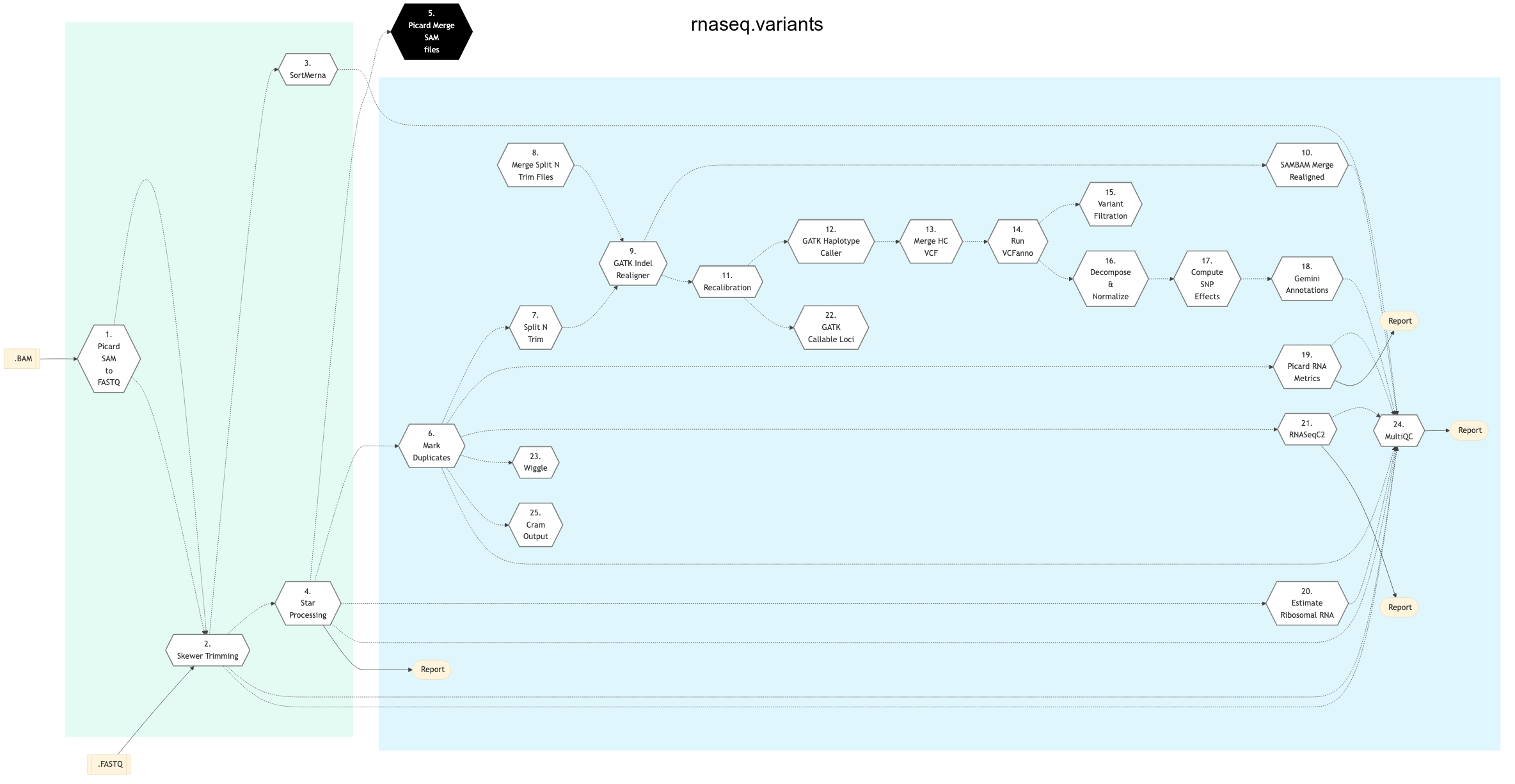
Figure: Schema of RNA Sequencing pipeline (variants)
Cancer
The following figure shows the schema of the RNA sequencing protocol (variants).
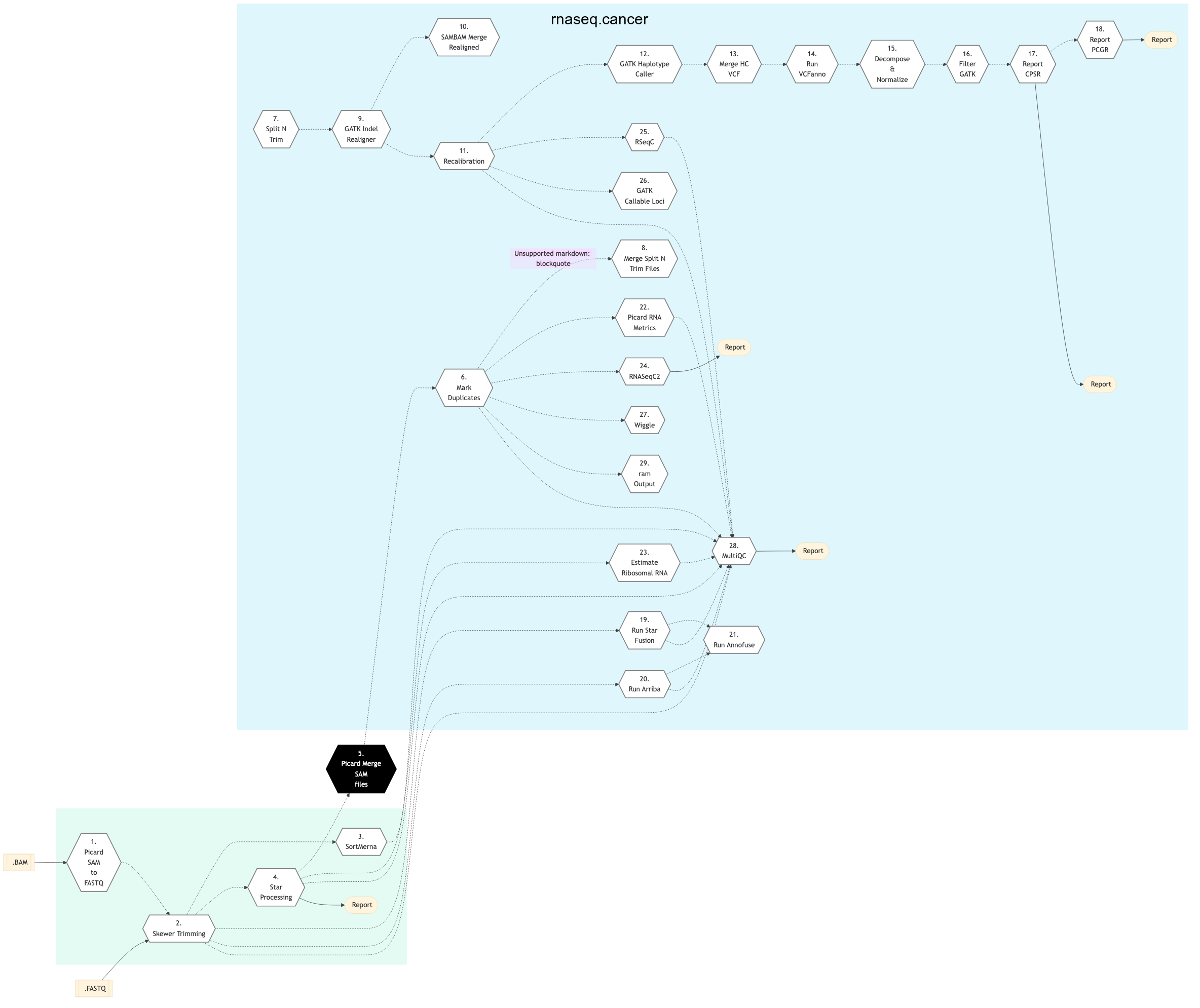
Figure: Schema of RNA Sequencing pipeline (cancer)
StringTie
Variants
Cancer
Picard SAM to FastQ
In this step, if FASTQ files are not already specified in the readset file, then it converts SAM/BAM files from the input readset file into FASTQ format. Otherwise, nothing is done.
Trimmomatic
This step takes as input files:
FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Raw reads quality trimming and removing of Illumina adapters is performed using Trimmomatic tool. If an adapter FASTA file is specified in the config file (section ‘trimmomatic’, param ‘adapter_fasta’), it is used first. Else, ‘Adapter1’ and ‘Adapter2’ columns from the readset file are used to create an adapter FASTA file, given then to Trimmomatic. For PAIRED_END readsets, readset adapters are reversed-complemented and swapped, to match Trimmomatic Palindrome strategy. For SINGLE_END readsets, only Adapter1 is used and left unchanged.
Merge Trimmomatic Stats
The trim statistics per readset are merged at this step.
Star Processing
The filtered reads are aligned to a reference genome. The alignment is done per readset of sequencing using the STAR software. It generates a Binary Alignment Map file (.bam).
This step takes as input files:
Trimmed FASTQ files if available
Else, FASTQ files from the readset file if available
Else, FASTQ output files from previous picard_sam_to_fastq conversion of BAM files
Run Star Fusion
STAR-Fusion is a component of the Trinity Cancer Transcriptome Analysis Toolkit (CTAT). Based on the STAR aligner it identifies candidate fusion transcripts supported by Illumina reads.
Picard Merge SAM Files
BAM readset files are merged into one file per sample. Merge is done using Picard Tool.
Picard Sort SAM
The alignment file is reordered (QueryName) using Picard Tool. The QueryName-sorted BAM files will be used to determine raw read counts.
Mark Duplicates
This step handles duplicates. Aligned reads per sample are duplicates if they have the same 5’ alignment positions (for both mates in the case of paired-end reads). All but the best pair (based on alignment score) will be marked as a duplicate in the BAM file. Marking duplicates is done using Picard package.
Picard RNA Metrics
Computes a series of quality control metrics using both CollectRnaSeqMetrics and CollectAlignmentSummaryMetrics functions metrics are collected using Picard package.
Estimate Ribosomal RNA
This step uses readset BAM files and bwa mem to align reads on the rRNA reference fasta and count the number of read mapped The filtered reads are aligned to a reference fasta file of ribosomal sequence. The alignment is done per sequencing readset. The alignment software used is BWA package with algorithm: bwa mem. BWA output BAM files are then sorted by coordinate using Picard package.
RNA Seq Compress
Computes a series of quality control metrics using RNA SeQC processing.
Wiggle
Generate wiggle tracks suitable for multiple browsers.
Raw Counts
Count reads in feature using HT Seq Count.
Raw Counts Metrics
Create rawcount matrix, zip the wiggle tracks and create the saturation plots based on standardized read counts.
Differential Expression
Performs differential gene expression analysis using DESEQ package and edgeR. Merge the results of the analysis in a single csv file.
StringTie Step
Assemble transcriptome using StringTie assembler.
StringTie Merge
Merge assemblies into a master transcriptome reference using StringTie assembler.
StringTie Abund
Assemble transcriptome and compute RNA-seq expression using StringTie.
Ballgown Gene Expression
Ballgown tool is used to calculate differential transcript and gene expression levels and test them for significant differences.
Sortmerna Step
This step calculates the ribosomal RNA per read based on known ribosomal sequences from archea, bacteria and eukaryotes. It uses sortmeRNA protocol that takes trimmed FASTQs and reports on each read, either paired-end or single end.
Rnaseqc2
Computes a series of quality control metrics using RNA-SeQC.
Skewer Trimming
Skewer is used mainly for detection and trimming adapter sequences from raw fastq files. Other features of Skewer is listed here.
Split N Trim
During the ‘Split N Trim’ step, a GATK Tool called SplitNCigarReads developed specially for RNAseq, splits reads into exon segments. During this splitting, it gets rid of Ns but maintains grouping information and hard-clips any sequences overhanging into the intronic regions.
Sambamba Merge Split N Trim Files
BAM readset files are merged into one file per sample. Merge is done using Sambamba Merge.
Sambamba Merge Realigned
In this step, BAM files of regions of realigned reads are merged per sample using Sambamba.
GATK Indel Realigner
Insertion and deletion realignment is performed on regions where multiple base mismatches are preferred over indels by the aligner since it can appear to be less costly by the algorithm. Such regions will introduce false positive variant calls which may be filtered out by realigning those regions properly. Realignment is done using GATK. The reference genome is divided by a number regions given by the nb_jobs parameter.
GATK Haplotype Caller
GATK haplotype caller step is used for SNPs and small indels. The Haplotype caller is capable of calling SNPs and indels simultaneously via local de-novo assembly of haplotypes in an active region. Regions that contain different types of variants close to each other are traditionally difficult to call. For such regions, HaplotypeCaller is more accurate. This is because whenever it encounters such regions with different types of variants, it discards the existing mapping information and completely reassembles the reads in that region.
GATK Callable Loci
This step computes the callable region or the genome as a BED track.
Filter GATK
As part of filter GATK processing, a custom script is applied to inject FORMAT information - tumor/normal DP and VAP into the INFO field of the filter on those generated fields.
Recalibration
In this step, we recalibrate the base quality scores of sequencing-by-synthesis reads in an aligned BAM file. After recalibration, the quality scores in the QUAL field in each read in the output BAM are more accurate in that the reported quality score is closer to its actual probability of mismatching the reference genome. Moreover, the recalibration tool attempts to correct for variation in quality with machine cycle and sequence context, and by doing so, provides not only more accurate quality scores but also more widely dispersed ones.
Merge HC VCF
Merges VCFs from Haplotype caller to generate a sample level VCF.
Run VCF Anno
VCFAnno is used to annotate VCF files with preferred INFO fields from any number of VCF or BED files.
Variant Filtration
VariantFiltration is a GATK tool for hard-filtering variant calls based on certain criteria. Records are hard-filtered by changing the value in the FILTER field to something other than PASS.
Decompose and Normalize
The vt Normalization is used to normalized and decompose VCF files. For more information about normalizing and decomposing visit Variant Normalization. An indexed file is also generated from the output file using htslib.
Compute SNP Effects
SnpEff is used to variant annotation and effect prediction on genes by using an interval forest approach. It annotates and predicts the effects of genetic variants, such as amino acid changes.
Gemini Annotations
Gemini (GEnome MINIng) is used to integrative exploration of genetic variation and genome annotations. See latest Gemini documentation for more information.
Report CPSR
In this step a cpsr germline report is created input: filtered ensemble germline vcf output: html report and additional flat files.
Report PCGR
Creates a PCGR somatic + germline report. Input is a filtered ensemble germline VCF and the output is an html report with additional flat files.
Run Arriba
Arriba is a command-line tool for the detection of gene fusions from RNA-Seq data. It is based on the STAR aligner. Apart from gene fusions, Arriba can detect other structural rearrangements with potential clinical relevance, including viral integration sites, internal tandem duplications, whole exon duplications and intragenic inversions, etc.
Run Annofuse
Annofuse is an R package used to annotate, prioritize, and interactively explore putative oncogenic RNA fusions.
RSeqC
RSeqC computes a series of quality control metrics using both CollectRnaSeqMetrics and CollectAlignmentSummaryMetrics functions metrics are collected using Picard.
Multiqc Report
A quality control report for all samples is generated. For more detailed information see MultiQC documentation.
CRAM Output
Generate long term storage version of the final alignment files in CRAM format. Using this function will include the original final BAM file into the removable file list.
The standard MUGQIC RNA-Seq pipeline has three protocols:
StringTie
Variants
Cancer
StringTie is the default protocol and applicable in most cases.
All three protocols are based on the use of the STAR aligner to align reads to the reference genome. These alignments are used during downstream analysis (for example in stringtie protocol, to determine differential expression of genes and transcripts).
The StringTie suite is used for differential transcript expression (DTE) analysis, whereas DESeq2 package and edgeR package are used for the differential gene expression (DGE) analysis.
StringTie protocol requires a design file which will be used to define the comparison groups in the differential analyses. Refer to the design file format. In addition, Ballgown package is used to calculate differential transcript and gene expression levels and test them for significant differences. It can also take a batch file (optional) which will be used to correct for batch effects in the differential analyses. Note the batch file format.
The variants protocol is used when variant detection, is the main goal of the analysis. GATK best practices workflow is used for variant calling. It also contains a step for annotating genes using the gemini protocol.
The cancer protocol contains all the steps in the variants protocol but it is specifically designed for cancer data sets due to the complexity of cancer samples and additional analyses those projects often entail. The goal of the cancer protocol is comparing expression to known benchmarks. In addition to the steps in the variants protocol, it contains four specific steps. Three of them (run_star_fusion, run_arriba, run_annofuse) are related to detection and annotation of gene fusions. For that, Star-fusion package, Arriba package and anno-Fuse package are used. Furthermore, RSeQC package provides RNA-seq quality control metrics to asses the quality of the data.
Finally, a summary html report is automatically generated by the pipeline at the end of the analysis. This report contains description of the sequencing experiment as well as a detailed presentation of the pipeline steps and results. Various Quality Control (QC) summary statistics are included in the report and additional QC analysis is accessible for download directly through the report. The report includes also the main references of the software tools and methods used during the analysis, together with the full list of parameters that have been passed to the pipeline main script.
See Schema tab for workflow. For the latest implementation and usage details refer to RNA Sequencing implementation README file file.
References