Long Read DNA Sequencing Pipeline
Version 6.1.0
Nanopore Protocol
The Long Read DNA Sequencing Pipeline introduced in GenPipes v6.x release supports three protocols:
Nanopore (A standalone pipeline in GenPipes v5.x and earlier)
Nanopore Paired Somatic New
Revio
Command
user@machine:~$ genpipes longread_dnaseq [-options ] [--genpipes_file GENPIPES_FILE.sh]
user@machine:~$ bash GENPIPES_FILE.sh
Options
-t {nanopore, nanopore_paired_somatic, revio},
--type {nanopore, nanopore_paired_somatic, revio}
Long Read DNA sequencing analysis type (default=nanopore)
-d DESIGN, --design DESIGN
design file
-r READSETS, --readsets READSETS
readset file
-h show this help message and exit
--help show detailed description of pipeline and steps
-c CONFIG [CONFIG ...],
--config CONFIG [CONFIG ...]
config INI-style list of files; config parameters
are overwritten based on files order
-s STEPS, --steps STEPS step range e.g. '1-5', '3,6,7', '2,4-8'
-o OUTPUT_DIR, --output-dir OUTPUT_DIR
output directory (default: current)
-j {pbs,batch,daemon,slurm},
--job-scheduler {pbs,batch,daemon,slurm}
job scheduler type (default: slurm)
-f, --force force creation of jobs even if up to date (default:
false)
Take the mem input in the ini file and force to have a
minimum of mem_per_cpu by correcting the number of cpu
(default: None)
--force_mem_per_cpu FORCE_MEM_PER_CPU
Take the mem input in the ini file and force to have a
minimum of mem_per_cpu by correcting the number of cpu
(default: None)
--json-pt create JSON file for project_tracking database
ingestion (default: false i.e. JSON file will NOT be
created)
- -report create 'pandoc' command to merge all job markdown
report files in the given step range into HTML, if
they exist; if --report is set, --job-scheduler,
--force, --clean options and job up-to-date status
are ignored (default: false)
--clean create 'rm' commands for all job removable files in
the given step range, if they exist; if --clean is
set, --job-scheduler, --force options and job up-to-
date status are ignored (default: false)
Note: Do not use -g option with --clean, use '>' to redirect
the output of the --clean command option
-l {debug,info,warning,error,critical},
--log {debug,info,warning,error,critical}
log level (default: info)
--sanity-check run the pipeline in `sanity check mode` to verify
all the input files needed for the pipeline to run
are available on the system (default: false)
--container {wrapper, singularity} <IMAGE PATH>
run pipeline inside a container providing a container
image path or accessible singularity hub path
--wrap [WRAP] path to the GenPipes cvmfs wrapper script (default:
genpipes/ressources/container/bin/container_wrapper.sh,
a convenience option for using GenPipes in a container)
-v, --version show the version information and exit
-g GENPIPES_FILE, --genpipes_file GENPIPES_FILE
Commands for running the pipeline are output to this
file pathname. The data specified to pipeline command
line is processed and pipeline run commands are
stored in GENPIPES_FILE, if this option is specified
. Otherwise, the output will be redirected to stdout
. This file can be used to actually "run the
GenPipes Pipeline"
Important
Do not use -g option with -clean.
Use ‘>’ to redirect the output of the genpipes command to a file when using -clean option.
Example
Nanopore
user@machine:~$ genpipes longread_dnaseq -t nanopore \ -c $GENPIPES_INIS/longread/longread.base.ini $GENPIPES_INIS/common_ini/rorqual.ini \ -r /cvmfs/soft.mugqic/root/testdata/nanopore/readset.nanopore.txt \ -o nanopore \ -g longread_nanopore_cmd.sh user@machine:~$ bash longread_nanopore_cmd.sh
Tip
Depending upon the cluster where you are executing the pipeline, substitute the file name rorqual.ini in the command with the appropriate <DRAC server cluster name>.ini file located in the $GENPIPES_INIS/common_ini folder.
For e.g., rorqual.ini, fir.ini, or narval.ini.
Nanopore Paired Somatic
Pair File
The long-read DNA sequencing pipeline nanopore paired somatic protocol requires a pair file as input using the -p option.
user@machine:~$ genpipes longread_dnaseq -t nanopore_paired_somatic \ -c $GENPIPES_INIS/longread/longread.base.ini \ $GENPIPES_INIS/common_ini/rorqual.ini \ $GENPIPES_INIS/longread/longread_dnaseq.cancer.ini \ -r /cvmfs/soft.mugqic/root/testdata/longread_dnaseq/readset.nanopore_paired_somatic.txt \ -p /cvmfs/soft.mugqic/root/testdata/longread_dnaseq/pairs.nanopore_paired_somatic.csv \ -o nanopore \ -g longread_nanopore_cmd.sh user@machine:~$ bash longread_nanopore_cmd.sh
Tip
Depending upon the cluster where you are executing the pipeline, substitute the file name rorqual.ini in the command with the appropriate <DRAC server cluster name>.ini file located in the $GENPIPES_INIS/common_ini folder.
For e.g., rorqual.ini, fir.ini, or narval.ini.
Revio
user@machine:~$ genpipes longread_dnaseq -t revio \ -c $GENPIPES_INIS/longread/longread.base.ini \ $GENPIPES_INIS/common_ini/rorqual.ini \ -r /cvmfs/soft.mugqic/root/testdata/longread_dnaseq/readset.revio.txt \ -o revio \ -g longread_revio_cmd.sh user@machine:~$ bash longread_revio_cmd.sh
Tip
Depending upon the cluster where you are executing the pipeline, substitute the file name rorqual.ini in the command with the appropriate <DRAC server cluster name>.ini file located in the $GENPIPES_INIS/common_ini folder.
For e.g., rorqual.ini, fir.ini, or narval.ini.
Caution
It is recommended that you use the -g GENPIPES_CMD.sh option instead of redirecting the output of the pipeline command to a file via > GENPIPES_CMD.sh.
user@machine:~$ genpipes [pipeline] [options] -g genpipes_cmd.sh
user@machine:~$ bash genpipes_cmd.sh
user@machine:~$ genpipes [pipeline] [options] > genpipes_cmd.sh
user@machine:~$ bash genpies_cmd.sh
The > scriptfile method is supported but will be deprecated in a future GenPipes release.
Use the Long Read DNA sequencing test dataset for this pipeline.
Nanopore
The following figure shows the schema for Nanopore sequencing pipeline:
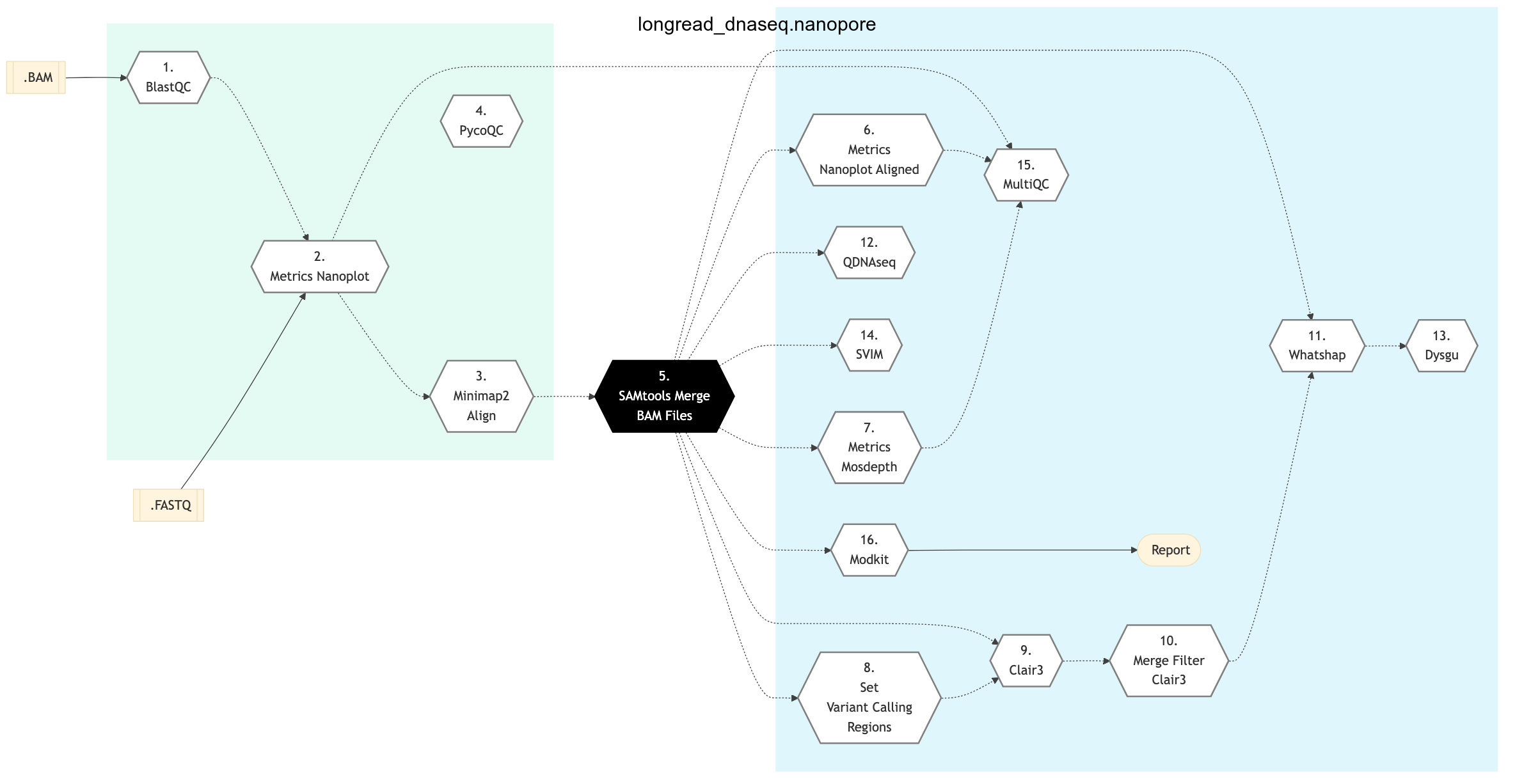
Figure: Schema of Nanopore Long Read DNA Sequencing Protocol
Nanopore Paired Somatic
The following figure shows the schema for Nanopore Paired Somatic sequencing pipeline:
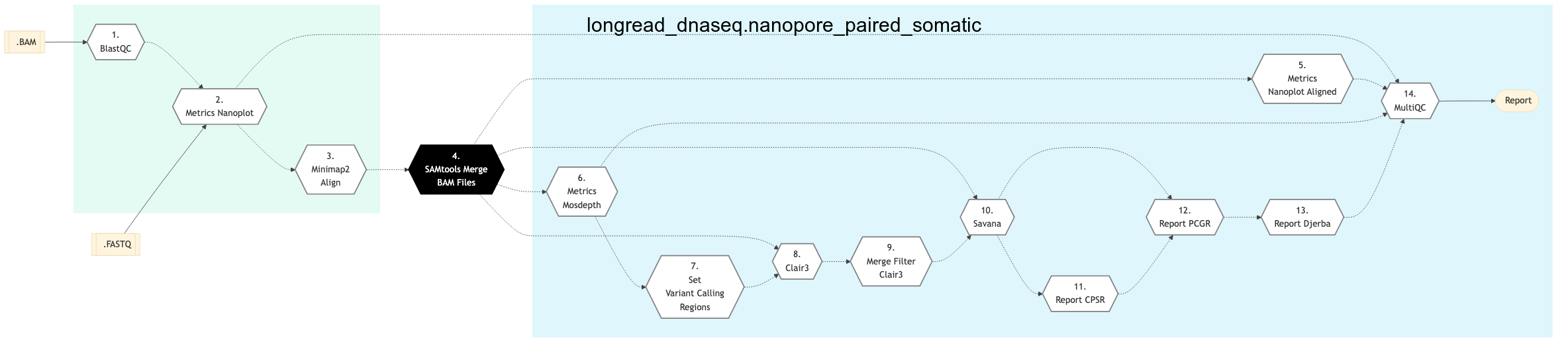
Figure: Schema of Long Read DNA Sequencing Nanopore Paired Somatic Protocol
Revio
The following figure shows the schema for Revio Long Read DNA Sequencing Protocol:
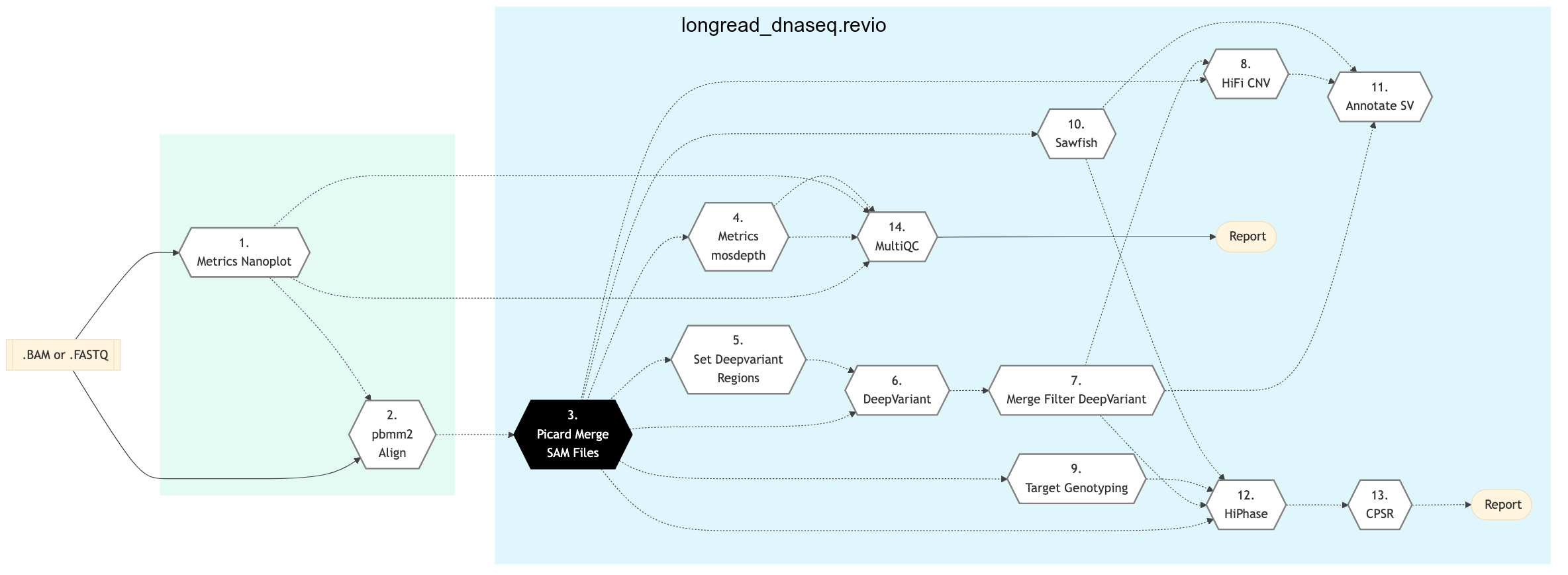
Figure: Schema of Nanopore Sequencing protocol
Nanopore
Nanopore Paired Somatic
Revio
BlastQC
In this step, Blast-QC utility is used for sequence alignment and identification. It performs a basic QC test by aligning 1000bp of randomly selected reads to the NCBI Nucleotide Database in order to detect potential contamination.
Minimap2 Align
Minimap2 Align Program is a fast, general purpose sequencing alignment program that maps DNA and long mRNA sequences against a large reference database. It can be used for Nanopore sequencing for mapping 1kb genomic reads at an error rate of 15% (e.g., PacBio or Oxford Nanopore genomic reads), among other uses.
In this step, minimap2 to align the FastQ reads that passed the minimum QC threshold to the provided reference genome. By default, it aligns to the human genome reference (GRCh38) with Minimap2.
SAMTools Merge BAM Files
BAM readset files are merged into one file per sample. Merge is done using Samtools.
This step takes as input files: Aligned and sorted BAM output files from previous minimap2_align or pbmm2_align step
pycoQC
In this step, pycoQC Software is used produce an interactive quality report based on the summary file and alignment outputs. PycoQC relies on the sequencing_summary.txt file generated by Guppy. If needed, it can also generate a summary file from basecalled FAST5 files. PycoQC computes metrics and generates interactive QC plots for Oxford Nanopore technologies sequencing data.
Whatshap
Create a haplo-tagged file using Whatshap.
QDNAseq
This step runs the QDNAseq R script.
Dysgu
Call structural variants with dysgu.
Picard Merge SAM Files
BAM readset files are merged into one file per sample in this step. Using aligned and sorted BAM output files from Minimap2 Align step, it performs the merge using Picard.
Structural Variant Identification using Mapped Long Reads
In this step, Structural Variant Identification using Mapped Long Reads (SVIM methodology), is used to perform structural variant (SV) analysis on each sample.
Metrics Nanoplot
This step collects QC metrics on unaligned BAM or FastQ files with Nanoplot.
Metrics Nanoplot Aligned
This step collects QC metrics on aligned BAM file with Nanoplot.
Create an interval list with ScatterIntervalsByNs from GATK. Used for creating a broken-up interval list that can be used for scattering a variant-calling pipeline in a way that will not cause problems at the edges of the intervals. By using large enough N blocks (so that the tools will not be able to anchor on both sides) we can be assured that the results of scattering and gathering the variants with the resulting interval list will be the same as calling with one large region.
pbmm2 Align
Uses pbmm2 to align fastq files or the raw HiFi BAM to the reference. Sorted output can be used directly for polishing using GenomicConsensus, if BAM has been used as input to pbmm2.
Metrics Mosdepth
Calculate depth stats with mosdepth.
Set DeepVariant Regions
Create an interval list with ScatterIntervalsByNs from GATK. Used for creating a broken-up interval list that can be used for scattering a variant-calling pipeline in a way that will not cause problems at the edges of the intervals. By using large enough N blocks (so that the tools will not be able to anchor on both sides) we can be assured that the results of scattering and gathering the variants with the resulting interval list will be the same as calling with one large region.
DeepVariant Germline VC
Germline variant calling with DeepVariant.
Merge Filter DeepVariant
Merge DeepVariant outputs from the previous step, if applicable, and filter vcf.
HiFi CNV
Call copy number variation and visualize results with HiFiCNV.
Target Genotyping
Call tandem repeats for pathogenic and full repeats with trgt for targeted genotyping.
Sawfish
Call structural variants from mapped HiFi sequencing reads with Sawfish SV Caller.
Annotate SV
Annotate and rank structural variants with AnnotSV.
Hi Phase
Phase variant calls with HiPhase.
Clair3 Step
Call germline small variants with clair3.
Merge Filter Clair3
Merge clair3 outputs, if applicable, and filter VCF.
Modkit Step
In this step methylation analysis is done for the Nanopore data.
Report CPSR
Creates a CPSR germline report. It takes annotated/filter VCF as the input and outputs an html report along with additional flat files.
Report PCGR
Creates a PCGR somatic plus a CPSR germline report with input as the filtered somatic VCF. The output is a html report and additional flat files.
Report Djerba
Creates a clinical report using Djerba and metadata along with workflow output.
Savana Step
Call somatic structural variants and copy number aberrations with Savana.
MultiQC
The MultiQC aggregator collates results from bioinformatics analyses across many samples into a single report. It searches for a given directory for analysis logs and compiles a HTML report. This is a tool for general usage, perfect for summarizing the output from numerous bioinformatics tools.
Over the past decade, long-read, single-molecule DNA sequencing technologies have emerged as powerful players in genomics. With the ability to generate reads tens to thousands of kilobases in length with an accuracy approaching that of short-read sequencing technologies, these platforms have proven their ability to resolve some of the most challenging regions of the human genome, detect previously inaccessible structural variants, and generate some of the first telomere-to-telomere assemblies of whole chromosomes.
The LongRead Pipeline is used to analyze long reads produced by the Oxford Nanopore Technologies (ONT) and PacBio Revio sequencers. It supports the following protocols:
Nanopore
Revio
Both protocols require a readset file as input. The readset file for the Long Read DNA Seq pipeline has a specific structure and format containing the sample metadata and paths to input data (FASTQ, FAST5 or BAM).
Nanopore
The Nanopore protocol of the pipeline uses minimap2 to align reads to the reference genome. Additionally, it produces a QC report that includes an interactive dashboard with data from the basecalling summary file as well as the alignment. A step aligning random reads to the NCBI nt database and reporting the species of the highest hits is also done as QC.
Once the QC and alignments have been produced, Picard is used to merge readsets coming from the same sample. Finally, SVIM is used to detect Structural Variants (SV) including deletions, insertions and translocations.
For a full summary of the types of SVs detected, refer to this site.
The SV calls produced by SVIM are saved as VCFs for each sample, which can then be used in downstream analyses. No filtering is performed on the SV calls.
This pipeline currently does not perform base calling and requires both FASTQ and a sequencing_summary file produced by a ONT supported basecaller (we recommend Guppy). Additionally, the testing and development of the pipeline were focused on genomics applications, and functionality has not been tested for transcriptomics or epigenomics datasets.
For more information on using ONT data for structural variant detection, as well as an alternative approach, refer to Structural Variant Pipeline GitHub repository.
Revio
The Revio protocol uses pbmm2 to align reads to the reference genome, followed by variant calling with DeepVariant and structural variant calling with HiFiCNV, TRGT, and Sawfish. Variants are annotated with AnnotSV and phased with HiPhase. A CPSR report can be produced from the phased variants. Metrics on the raw and mapped reads are collected with NanoPlot and mosdepth, respectively.
See Schema tab for the pipeline workflow. For the latest implementation and usage details refer to the Long Read DNA Sequencing implementation README.md file.
References
Evaluating nanopore sequencing data processing pipelines for structural variation identification.
Minimap2: Pairwise alignment for nucleotide sequences.