Introducing GenPipes¶
GenPipes is a workflow management system (WMS) that facilitates building robust genomic workflows. It is a unique solution that combines both a framework for development and end-to-end analysis pipelines, covering a large scope of genomic applications and research domains. It offers researchers a simple method to analyze different types of data, customizable to their needs and resources. In addition, it provides flexibility to create customized workflows besides the ones that are already implemented and validated by GenPipes.
What is GenPipes?¶
GenPipes is an open source, flexible, scalable Python-based framework that facilitates the development and deployment of multi-step computational workflows. These workflows are optimized for High-Performance Computing (HPC) clusters and the cloud.
Following genomics application pipelines are already implemented and validated through GenPipes:
ChIP-Seq
De-Novo RNA Sequencing
Deep Whole Genome Sequencing
Exome Sequencing
Hi-C / Capture Hi-C
Illumina raw data processing
Metagenomics
SARS-CoV-2 genome sequencing
RNA Sequencing / Unmapped RNA Quality Control
Transcriptonomics Assembly
Tumour Analysis
Whole Exome Sequencing (WES)
Whole Genome Bisulphate Sequencing (WGBS)/ Reduced Representation Bisulphate Sequencing (RRBS)
Whole Genome Sequencing (WGS)
GenPipes software is available under a GPLv3 open source license and is continuously updated to follow recent advances in genomics and bioinformatics. The framework can be accessed through multiple deployment mechanisms. It has already been configured on several servers at C3G HPC computing facility. Its also supports Cloud deployment through GCP. Besides this, a Docker image is also available to facilitate additional installations on local / individual machines for small dataset analysis.
Basic Concepts¶
GenPipes is a Python based object oriented workflow management system that comes pre-built with several genomic analysis pipelines. A typical analysis workflow comprises of several complex actions that are interdependent and need to be managed in terms of input, output, process configuration and pipeline tuning.
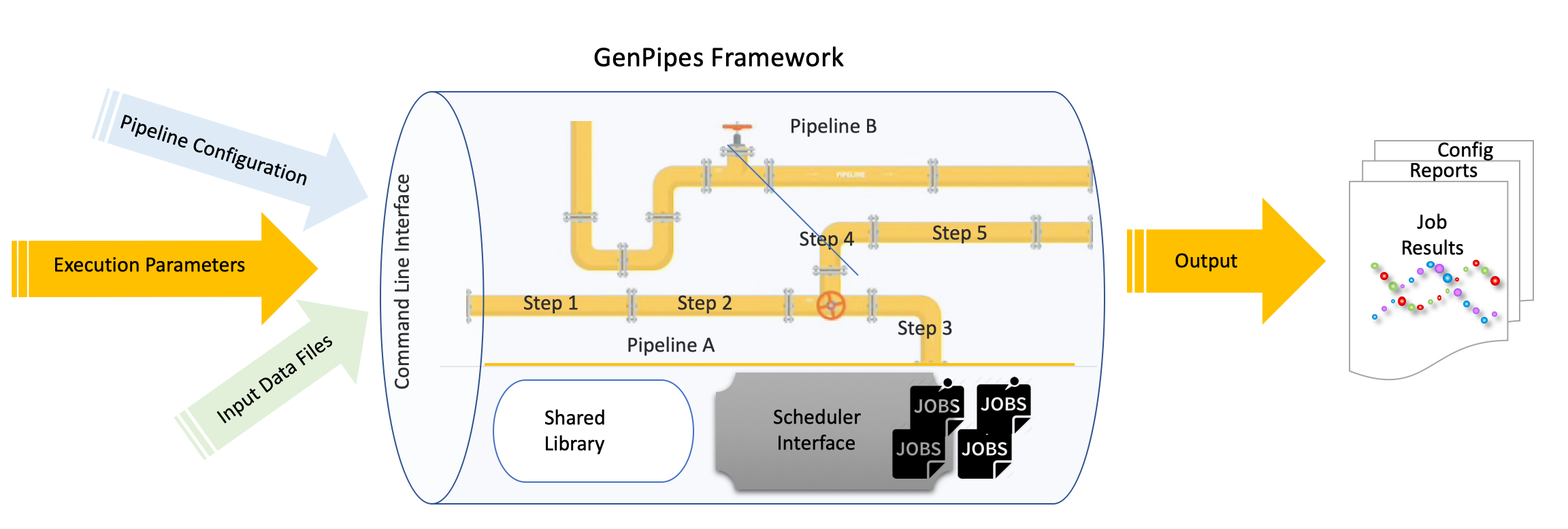
GenPipes refer to four kinds of objects that are used to manage different components of a typical analysis workflow. These are:
Pipeline
Step
Job
Scheduler
Pipeline¶
It is the main object that controls the overall genomic analysis workflow. For each type of analysis, a specific Pipeline object is defined. Pipeline objects can inherit from one another.
Step¶
Each Pipeline object uses Step objects to define the flow of the analysis. It provides flexibility in choosing which steps are called during a pipeline execution. Pipeline instance calls all steps implemented in a pipeline or only a set of steps selected by the user. Each step of a pipeline is a unit of execution block that encapsulates a part of the analysis (e.g., trimming or alignment). The Step object is a central unit object that corresponds to a specific analysis task. The execution of the task is directly managed by the code defined in each Step instance; some steps may execute their task on each sample individually while other steps execute their task using all the samples collectively.
Job¶
The key purpose of each Step object is to generate a list of “Job” objects, which correspond to the consecutive execution of single tasks. The Job object defines the commands that will be submitted to the system. It contains all the elements needed to execute the commands, such as input files, modules to be loaded, as well as job dependencies and temporary files. Jobs are submitted in a workflow management system which uses various algorithms to schedule various types of jobs and optimizes time to result and resource usage.
Scheduler¶
Each Job object is submitted to the GenPipes workflow management system using a specific “Scheduler” object. The Scheduler object creates execution commands that are compatible with the user’s computing system. Four different Scheduler objects have already been implemented (PBS, SLURM, Batch, and Daemon).
PBS scheduler creates a batch script that is compatible with a PBS (TORQUE) system.
SLURM scheduler creates a batch script that is compatible with a SLURM system.
Batch scheduler creates a batch script that contains all the instructions to run all the jobs one after the other.
Daemon scheduler creates a log of the pipeline command in a JSON file.
How GenPipes works?¶
GenPipes is a Python based object-oriented framework that is available to users as a command line tool. Figure below shows the general workflow of GenPipes.
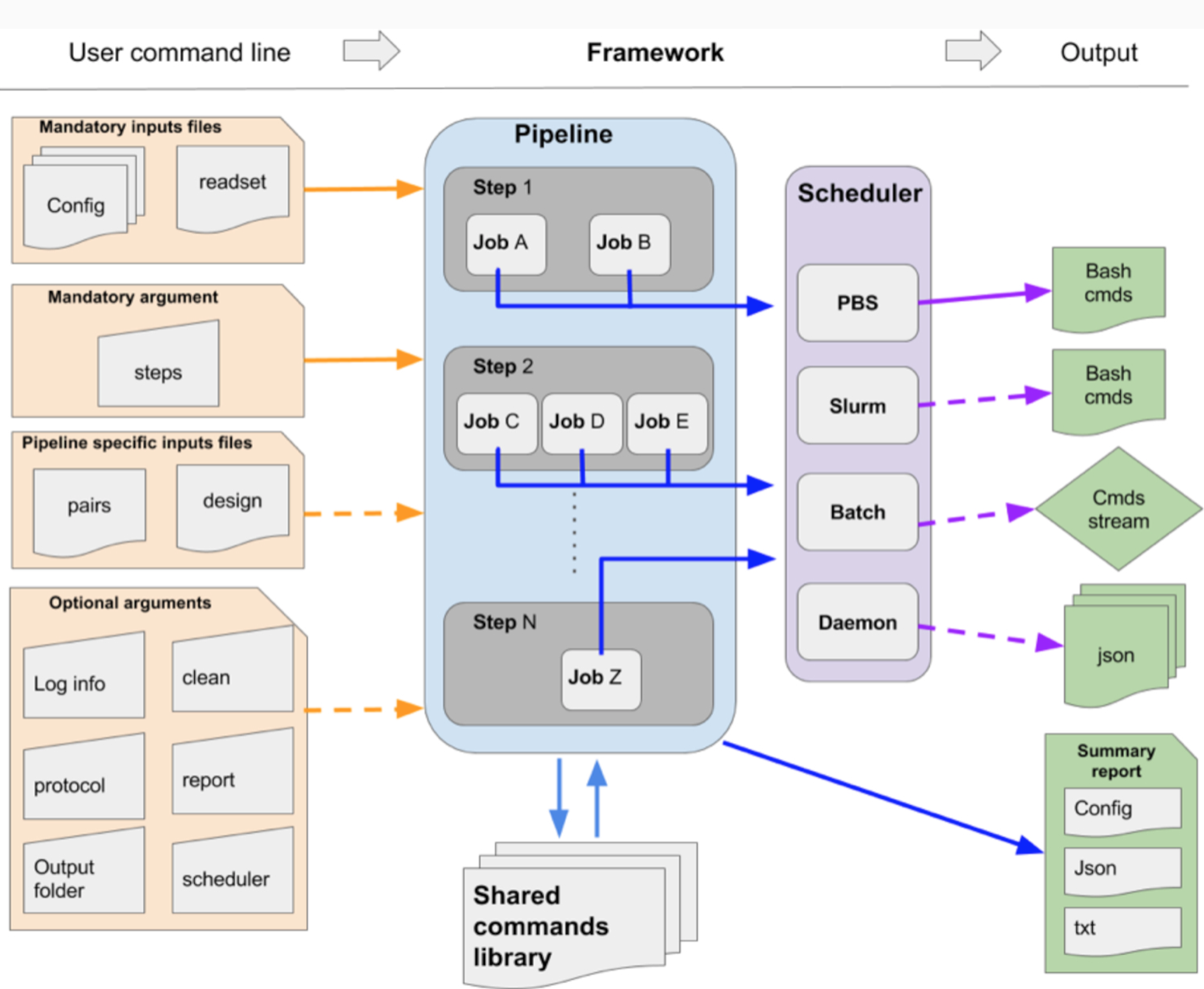
Each GenPipes Pipeline can be launched using a command line instruction. There are three kinds of inputs for each such instruction as shown in the figure above.
Mandatory command options
Optional command options
Input Files
The input files can be of two kinds - mandatory ones, that are needed for every pipeline and pipeline specific input files.
Mandatory input files include Configuration files and Readset files. Configuration files contain details regarding machine environment where the pipeline is executed and parameters that need to be set for each step of the pipeline. Default values are provided and can be changed in case of specific genomic analysis. GenPipes can be deployed locally in your data center or users can access pre-installed GenPipes on Compute Canada servers. For details regarding different kinds of GenPipes deployment, refer to GenPipes Deployment Guide. If you are using GenPipes pre-installed on Compute Canada servers, then the basic configuration files are installed along with GenPipes. These can be supplemented with additional configuration files provided using the ‘-c’ option while running the command line instruction.
Besides the mandatory configuration files, some pipelines have their own specific input file that must be provided. These include Design Files and Test Dataset files. These files are not provided by default and users need to supply them while running the pipelines. For the pipelines that require test dataset files, if you do not have access to any test datasets, you can try out some of the available Sample Test Dataset Files that are available as additional GenPipes resources for users.
When the GenPipes command is launched, required modules and files will be searched for and validated. If all required modules and files are found, the analysis commands will be produced. GenPipes will create a directed acyclic graph that defines job dependency based on input and output of each step.
Once launched, the jobs are sent to the scheduler and queued. As jobs complete successfully, their dependent jobs are released by the scheduler to run. If a job fails, all its dependent jobs are terminated and an email notification is sent to the user. When GenPipes is re-run, it will detect which steps have successfully completed, as described in section “Smart relaunch features,” and skip them but will create the command script for the jobs that were not completed successfully. To force the entire command generation, despite successful completion, the “-f” option should be added. The output of the command line instruction are in the form of summary reports and job status. Depending upon the pipeline, there are varied tools that can be used to view and analyze the results. See Viewing and Analyzing GenPipes Results for further information.
For details on GenPipes usage and various bioinformatics pipelines see GenPipes User Guide.
Bioinformatics and the role of GenPipes¶
There has been significant technological evolution in Next Generation Sequencing techniques from improvement in the processes themselves, better infrastructure and software availability as well as in terms of lowering of costs associated with NGS processing. For a good primer on the topic, refer to Introduction to Next Generation Sequencing.
GenPipes plays a key role in data pipelining and processing of next generation sequencing data and cutting edge genomic analysis, as highlighted in the figure below: