Running GenPipes
This section provides instructions on setting up GenPipes execution environment and using the available pipelines for genomic analysis. It assumes you already have access to GenPipes through one of the available deployment options.
GenPipes Executable
GenPipes enables various genomic analyses using its pipelines. It is a command-line tool built using a Python-based, object-oriented framework. This framework simplifies developing new features and adapting to new systems. New genomic workflows can be added in the framework by implementing a Pipeline object that inherits features and steps from existing objects.
Deploying GenPipes on a new system requires developing a Scheduler object and system specific configuration files. GenPipes’s command execution uses a shared library system, enabling task modification through input parameter adjustments. This approach simplifies code maintenance and ensures consistent software version updates across all pipelines.
Usage
Prerequisites
Before running GenPipes, review the checklist of prerequisites.
To launch GenPipes, use the following command:
user@machine:~$ genpipes <pipeline-name> -c config -r readset-file -s 1-n -g list-of-commands.txt
Then, execute the generated script:
user@machine:~$ bash list-of-commands.txt
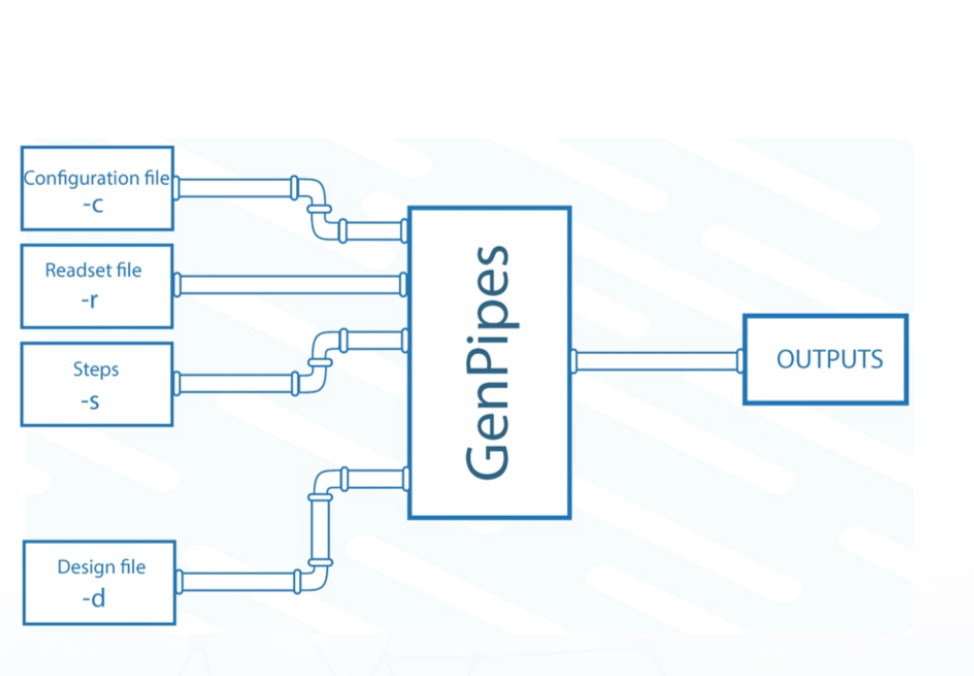
Command Inputs
Pipeline: Name of the pipeline. See Pipeline Reference Guide latest information on available pipelines and supported protocols.
Readset File: A readset file contains input about the samples. It is specified with the
-rflag. GenPipes aggregates and merges samples as per the readset file.Configuration File: A Configuration file or the
.inifile contains input parameters related to the cluster and the third-party tools used in the genomic analysis. It is specified with-cflag. Multiple configuration files can be specified for a cluster.Design File: A design file is required by some pipelines such as the
chipseqandrnaseq. This is in addition to the configuration or.inifile and the readset file. The design file specifies sample contrasts and groupings. It is specified with-dflag.Test Dataset: The Test Datasets could be simulated or real data generated by genomic analysis instruments. It is measured, sampled and read into various specified bioinformatics data formats and supplied as input to the pipelines.
Steps: The specific steps to be executed are indicated by the
-sflag. By default, all the pipeline steps are executed.Command File: The
-goption specifies the output command file name. This output file is used to finally launch the pipeline with the specified inputs.
Example Run
In this example, we will run the chipseq pipeline on the “Rorqual" server in Digital Research Alliance of Canada (DRAC). It requires you to download the ChiP Sequencing Test Dataset. The test dataset .tar file contains rawData folder with
FASTQ read files and readset file, readset.chipseq.txt, that describes that dataset.
user@machine:~$ genpipes chipseq -c $GENPIPES_INIS/chipseq/chipseq.base.ini \ $GENPIPES_INIS/common_ini/rorqual.ini \ -r readset.chipseq.txt \ -s 1-15 \ -g chipseq_cmd.sh
To understand what $GENPIPES_INIS refers to, please see instructions on how to access GenPipes on Compute Canada servers.
In the command above,
-cdefines the ini configuration files-rdefines the readset file-sdefines the steps of the pipeline to execute
Use genpipes chipseq -h to check details on various steps in the pipeline.
By default, Slurm scheduler is used when using the GenPipes deployment on the DRAC servers such as Rorqual, Nibi, Fir, Trillium and Narval. The abacus server uses the PBS scheduler. For that you need to specify “-j pbs” option as shown below:
user@machine:~$ genpipes chipseq -c $GENPIPES_INIS/chipseq/chipseq.base.ini \ $GENPIPES_INIS/common_ini/abacus.ini \ -r readset.chipseq.txt \ -s 1-15 \ -j pbs \ -g chipseq_cmd.sh
The above command generates a list of instructions that need to be executed to run the ChIP sequencing pipeline. These instructions are stored in the file chipseq_cmd.sh
To execute these instructions, use:
user@machine:~$ bash chipseq_cmd.sh
Warning
You will not see anything happen, but the commands will be sent to the server job queue. So do not run this more than once per job.
To confirm that the commands have been submitted, wait a minute or two depending on the server and type:
user@machine:~$ squeue -u <userID>
where, <userID> is your login id for accessing the DRAC infrastructure.
On abacus, the equivalent command is:
user@machine:~$ showq -u <userID>
In case you ran the command to submit the jobs several times and launched too many commands you do not want, you can use the following line of code to cancel ALL commands:
user@machine:~$ scancel -u <userID>
To cancel on abacus using PBS scheduler, use the command:
user@machine:~$ showq -u <userID> | tr "|" " "| awk '{print $1}' | xargs -n1 canceljob
After the processing is complete, you can access quality control plots in the report/ directory and you can find peak data in the peak_call/ directory.
For more information about output formats please consult the webpage of the third party tools used in the pipeline steps. See Pipeline Reference Guide for pipeline steps and third party tools usage.
Note
The ChIP sequencing pipeline also analyzes ATAC-Seq data if the “-t atacseq” flag is used. For more information on the available steps in that pipeline use:
user@machine:~$ genpipes chipseq -h
Example Run With Design File
Certain pipelines that involve comparing and contrasting samples, need a Design File. The design file can contain more than one way to contrast and compare samples. To see how this works with GenPipes pipelines, lets run a RNA-Sequencing experiment.
Step 1: Download the RNA-Sequencing Test Dataset.
The test dataset consists of the following files in the folder rawData:
FASTQ read files
Readset file
readset.rnaseq.txtDesign file
design.rnaseq.txt
The readset.rnaseq.txt file has the following contents:
Sample Readset Library RunType Run Lane Adapter1 Adapter2 QualityOffset BED FASTQ1 FASTQ2 BAM
GM12878_Rep1 GM12878_Rep1 myLibrary PAIRED_END 1 1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT 33 raw_data/rnaseq_GM12878_chr19_Rep1_R1.fastq.gz raw_data/rnaseq_GM12878_chr19_Rep1_R2.fastq.gz
GM12878_Rep2 GM12878_Rep2 myLibrary PAIRED_END 1 1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT 33 raw_data/rnaseq_GM12878_chr19_Rep2_R1.fastq.gz raw_data/rnaseq_GM12878_chr19_Rep2_R2.fastq.gz
H1ESC_Rep1 H1ESC_Rep1 myLibrary2 PAIRED_END 1 1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT 33 raw_data/rnaseq_H1ESC_chr19_Rep1_R1.fastq.gz raw_data/rnaseq_H1ESC_chr19_Rep1_R2.fastq.gz
H1ESC_Rep2 H1ESC_Rep2 myLibrary2 PAIRED_END 1 1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT 33 raw_data/rnaseq_H1ESC_chr19_Rep2_R1.fastq.gz raw_data/rnaseq_H1ESC_chr19_Rep2_R2.fastq.gz
There are four samples in this file with a single readset per sample. They are all PAIRED_END runs and have a pair of FASTQ
files located in the same rawData folder.
The design.rnaseq.txt file has the following contents:
Sample H1ESC_GM12787
H1ESC_Rep1 1
H1ESC_Rep2 1
GM12878_Rep1 2
GM12878_Rep2 2
The design file above contains a single analysis that compares two replicates of H1ESC to two replicates of group GM12878.
Step 2: Next, we will run this RNA-Sequencing analysis on the Rorqual server at Digital Research Alliance of Canada (DRAC), formerly Compute Canada (CCDB).
Use the following command to set up the pipeline:
user@machine:~$ genpipes rnaseq -c $GENPIPES_INIS/rnaseq/rnaseq.base.ini \ $GENPIPES_INIS/common_ini/rorqual.ini \ -r readset.rnaseq.txt \ -d design.rnaseq.txt \ -g rnaseqScript.txt\
Launch the pipeline via this command:
user@machine:~$ bash rnaseqScript.txt
Step 2: Finally, check launch status. The commands will be sent to the job queue and you will be notified once each step is done. If everything runs smoothly, you will see the output as MUGQICexitStatus:0 or Exit_status=0. In case an error occurs, the pipeline aborts. To examine the errors, check the content of the job_output folder.
Utilities to Manage Scheduling
HPC site policies typically limit the number of jobs that a user can submit in a queue. These sites deploy resource schedulers such as Slurm, or PBS/Torque for scheduling and sharing of HPC resources. Integrating with the resource schedulers and dealing with resource constraints are critical to ensuring productivity of HPC users.
GenPipes caters to these user pain points through intelligent tools and utilities that integrate with the schedulers. They help in smartly chunking the jobs, submitting pipeline runs, resubmitting the jobs when required and ensuring that there are no errors in scheduler calls. For a complete list of available GenPipes utilities, refer to the genpipes/util folder in the source tree.
Chunking Jobs
Use GenPipes tools `chunk_genpipes.sh` and `submit_genpipes` to enable better integration with resource schedulers (Slurm, PBS/Torque) deployed on HPC clusters. The `chunk_genpipes.sh` script is used to create job chunks of specified size that are submitted at a time.
Chunk once
`chunk_genpipes`script should be executed only once before issuing`submit_genpipes`to submit chunked jobs.
Submitting Job Chunks
The `submit_genpipes` script allows delegating job submission and managing resource constraints in a flexible and robust manner. It comes with a fail safe mechanism that will resubmit jobs that failed to be sent to the scheduler up to 10 times (default). There is an intelligent lock mechanism that prevents invoking two simultaneous runs of `submit_genpipes` in parallel, on the on the same job chunking folder or GenPipes pipeline run.
Submitting Chunks
The `submit_genpipes` script can be run for multiple GenPipes pipelines simultaneously, and submit jobs belonging to respective pipelines. You need to ensure that each submit_genpipes script invocation refers to a different job_chunks folder corresponding to the pipeline. The script runs can be stopped by `Ctrl-C` keystroke and restarted at will.
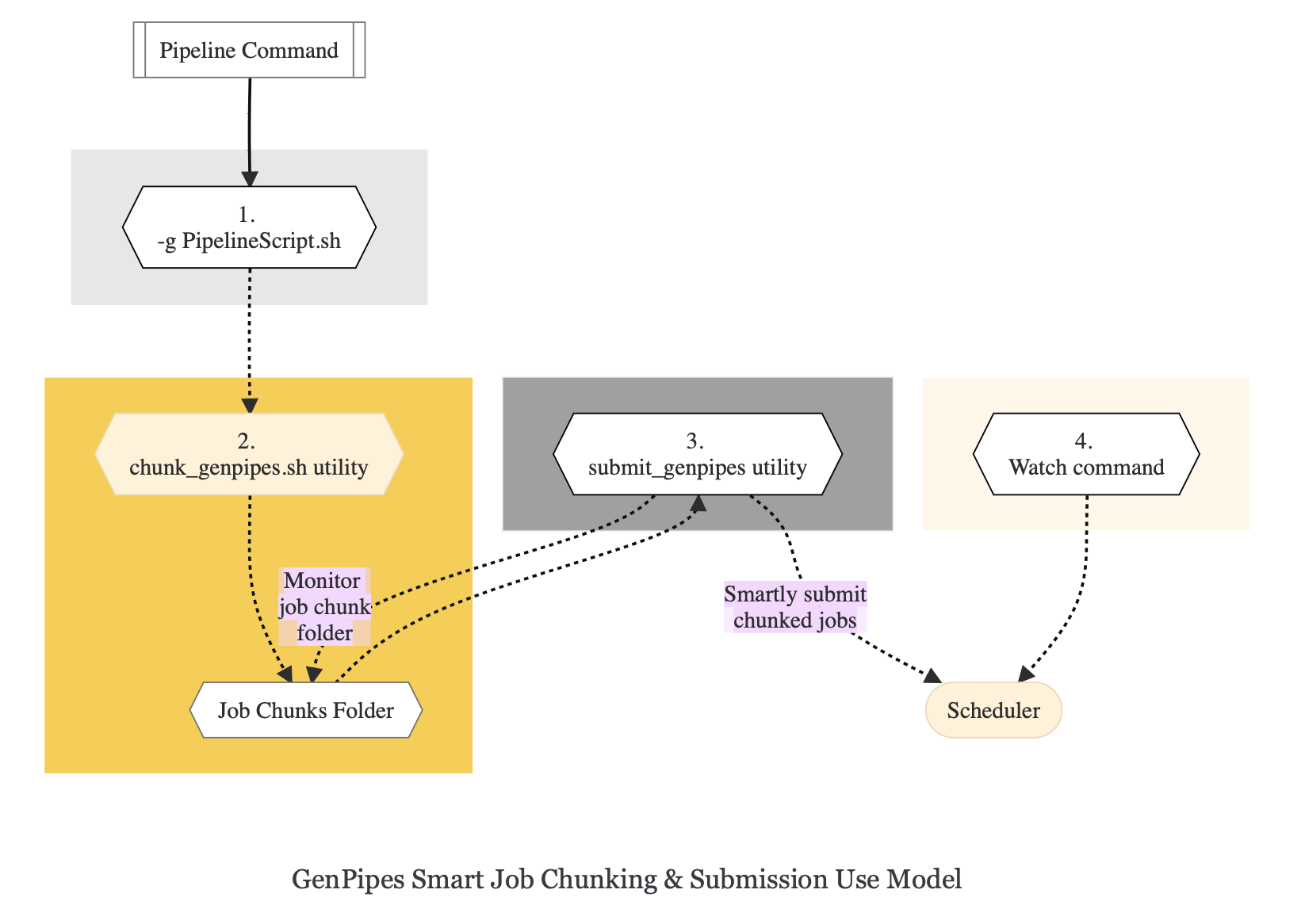
Use Model
Issue GenPipes pipeline command with -g GENPIPES_FILE option and store all the output pipeline commands in a bash script.
Provide this bash script as input to the
`chunk_genpipes.sh`tool to create scheduler job chunks and store into a folder`job_chunks`(default) or the one you specify. Note that chunk_genpipes.sh utility is supposed to be run for a pipeline bash script only once.After successful chunking, use
`submit_genpipes`tool to smartly submit the pipeline jobs to the scheduler. It takes care of scheduler integration, managing queue limits, and checking for errors in the calls made to the scheduler. makes sure to auto-correct them based on chunking limits specified through`chunk_genpipes.sh`earlier.Use
`watch`command to monitor the submitted jobs.At any time, you can stop monitoring the submitted jobs by issuing
`Ctrl-C`to a running`watch`command in the terminal.If the watch command is stopped cleanly via a
`ctrl-C`or killed in another manner, for example, when a session is killed after ssh disconnection, you can restart monitoring GenPipes jobs to the queuing system by invoking the`watch`command again.
The figure below demonstrates how the `submit_genpipes` utility works. The pipeline command file output is fed into `chunk_genpipes.sh` script which creates the chunks folder as a one time activity. This chunk folder is monitored by the `submit_genpipes` script.
Chunking Example
This example uses the Chip Sequencing Pipeline to show how to chunk GenPipes pipeline jobs, submit them, and monitor job status using the following utilities and commands:
chunk_genpipes.shsubmit_genpipeswatch
Step A: Construct the GenPipes command
Use the -g option to create the output command file for GenPipes chipseq pipeline, say chipseq_script.sh.
user@machine:~$ export M_FOLDER=path_to_folder
user@machine:~$ genpipes chipseq <options> --genpipes_file chipseq_script.sh
user@machine:~$ chunk_genpipes.sh chipseq_script.sh $M_FOLDER
user@machine:~$ submit_genpipes $M_FOLDER
Step B: Chunk Pipeline Commands
Use chunk_genpipes to the scheduler and specify the dnaseq.sh file as input. In the command below,
20 specifies the number of jobs in a chunk.
user@machine:~$ chunk_genpipes.sh chipseq_script.sh job_chunks 20
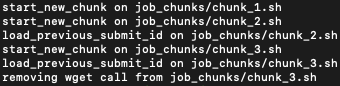
Fig: Output of chunk_genpipes command
Step C: Submit Job Chunks
Invoke submit_genpipes script to submit the job chunks for processing. Use watch to monitor the
submitted job chunks. The value 800 in the submit command below refers to the total number of jobs that
can be submitted simultaneously at a time to the scheduler.
user@machine:~$ submit_genpipes job_chunks -n 800
Figure below shows the output of the submit_genpipes command:
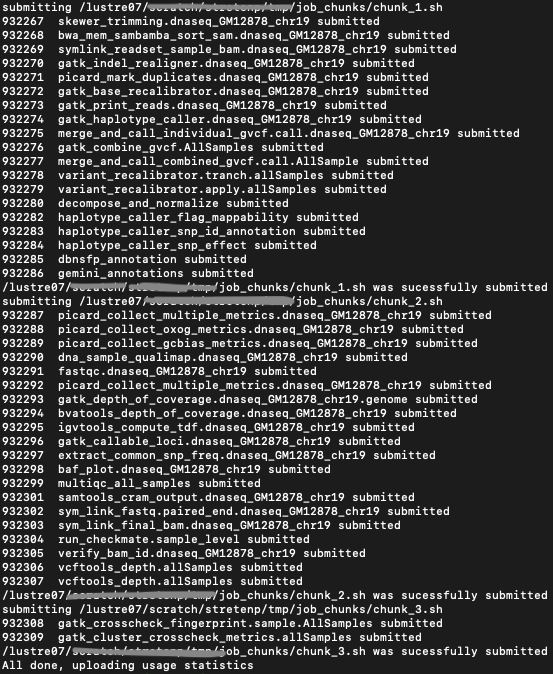
Fig: Output of the submit_genpipes command
Further Information
GenPipes pipelines are built around third party tools that the community uses in particular fields. To understand the output of each pipeline, please read the documentation pertaining to the tools that produced the output.
To learn more on how to run GenPipes, see GenPipes Tutorials. Refer to the list of available GenPipes pipelines for details on which pipelines and protocols are supported in the latest release.
If you have any further queries, drop us an email.