Troubleshooting Guide
No jobs are submitted after running genpipes command. Why?
The genpipes command simply spits out the list of jobs that will be submitted subsequently. These commands are stored in the genpipes_cmd.sh script. It does not run the jobs on its own, users must run this script to ensure jobs are submitted.
New users sometimes don’t realize that it is not sufficient to issue the genpipes <pipeline> [options] -g genpipes_cmd.sh command. You must also run bash genpipes_cmd.sh` after running the genpipes` command. Jobs are submitted only when you execute the bash genpipes_cmd.sh.
Issues while running genpipes command.
Often pipeline users encounter issues are due to the readset and/or design file not being formatted correctly. Make sure you use the correct format for the readset and the design file as mentioned in the pipeline user guide. For example, ChIP sequencing protocol uses a different file format than the DNA sequencing pipeline. Learn more about the different design file and readset file formats.
Why am I seeing low mapping rates?
Users often see very low mapping rates (close to 0%). This can be usually resolved by flipping the R1 and R2 reads. Use R2=FASTQ1 and R1=FASTQ2.
Why do I see this error - `qsub command not found`?
While running a pipeline using specific configuration file, the command text file is generated successfully. However, when the user tries to run the commands using:
user@machine:~$ bash <command-file.txt>
the following error shows up:
qsub: command not found

Fix
Check the kind of GenPipes deployment you are working with. If it is a local deployment, on a bare metal server or a virtual server or in a container, you need to make sure you do not specify -j pbs or -j slurm option but -j daemon or -j batch mode.
In case you are using Moab or Torque scheduler, ensure that you use -j pbs option in the pipeline command. If you are using mp2b or Rorqual server, then you need to specify -j slurm as the scheduler option and use the correct configuration file (mp2b.ini or key_ccdb_server_ini_name) depending upon which server you are using to run your pipeline jobs.
Fatal Limit Error: Star RNA Sequencing
When user issues the RNA sequencing GenPipes pipeline with Star option commands, the jobs fails with the fatal limit error:
Fatal LIMIT error: the number of junctions to be inserted on the fly =2663181 is larger than the limitSjdbInsertNsj=1000000 Fatal LIMIT error: the number of junctions to be inserted on the fly =2663181 is larger than the limitSjdbInsertNsj=1000000 SOLUTION: re-run with at least --limitSjdbInsertNsj 2663181 Nov 29 14:10:58 ...... FATAL ERROR, exiting MUGQICexitStatus:104
It is not clear from the error message where this solution configuration option needs to be specified.
Typically, the Star index options in the `.ini` file supplied for RNA sequencing protocol do not show `--limitSjdbInsertNsj` option.
[star_index] ram = 191000M io_buffer = 1G threads = 20 cluster_cpu = -N 1 -c 40 cluster_walltime = --time=15:00:0 cluster_queue = --mem-per-cpu=4775M star_cycle_number = 99
Fix
The correct way to specify this option is using `--other-option` flag as shown in the snippet from `.ini` file below:
[star_index]
ram = 191000M
io_buffer = 1G
threads = 20
cluster_cpu = -N 1 -c 40
cluster_walltime = --time=15:00:0
cluster_queue = --mem-per-cpu=4775M
star_cycle_number = 99
other_options =--limitSjdbInsertNsj 2500000
Error: Missing Genomes and Annotations
Several users have encountered this issue whereby genomes and annotations are missing after pipeline runs.
Fix
Most of the GenPipes pipeline commands require you to supply input data in the form of readsets, design files and configuration. Refer to the GenPipes Test Datasets and reference genomes, if a specific genome that you need to provide to the pipeline is not available in the GenPipes deployment on the Digital Research Alliance of Canada (DRAC),formerly Compute Canada, servers.
Why does the pipeline does not execute at all?
First time users may issue the pipeline command and assume it will generate jobs on worker nodes automatically. However, after multiple runs, no execution happens if the pipeline command is executed. For example see Han’s issue in the GenPipes Google Group.
Fix
This is a very common issue. GenPipes pipeline command does NOT issue the commands on its own. When you run the pipeline, it simply generates a bunch of commands to execute but does not execute them. You need to redirect the output of pipeline command into a file and then bash execute that file containing all the commands corresponding to a genomic analysis. See GenPipes Google Group discussions and Mathieu Bourgey’s response for details.
Out of Memory error in RNA Sequencing Star alignment
For first time users, it has been observed (see example in Google GenPipes Forum) that the RNA Sequencing pipeline command execution stops after STAR alignment 1.
Fix
Try to change the STAR parameters in your ini files to something like in the .ini files of the branch specific to the release:
https://github.com/c3g/GenPipes/tree/main/genpipes/pipelines/common_ini
https://bitbucket.org/mugqic/mugqic_pipelines/src/master
The problem should be solved by setting io_buffer to a higher value like 1G or 4G. The command you show indicates you are using 8M.
At some point io_buffer was decreased in the template .ini but this exposed a bug in STAR where a negative memory allocation is attempted.
Error: RAP_ID not set
If you try to run GenPipes deployed by C3G on the Digital Research Alliance of Canada (DRAC), formerly Compute Canada, servers, the initial run shows error related to RAP_ID not set. Sometimes, this same issue manifests in the form of timing error as shown in figure below:
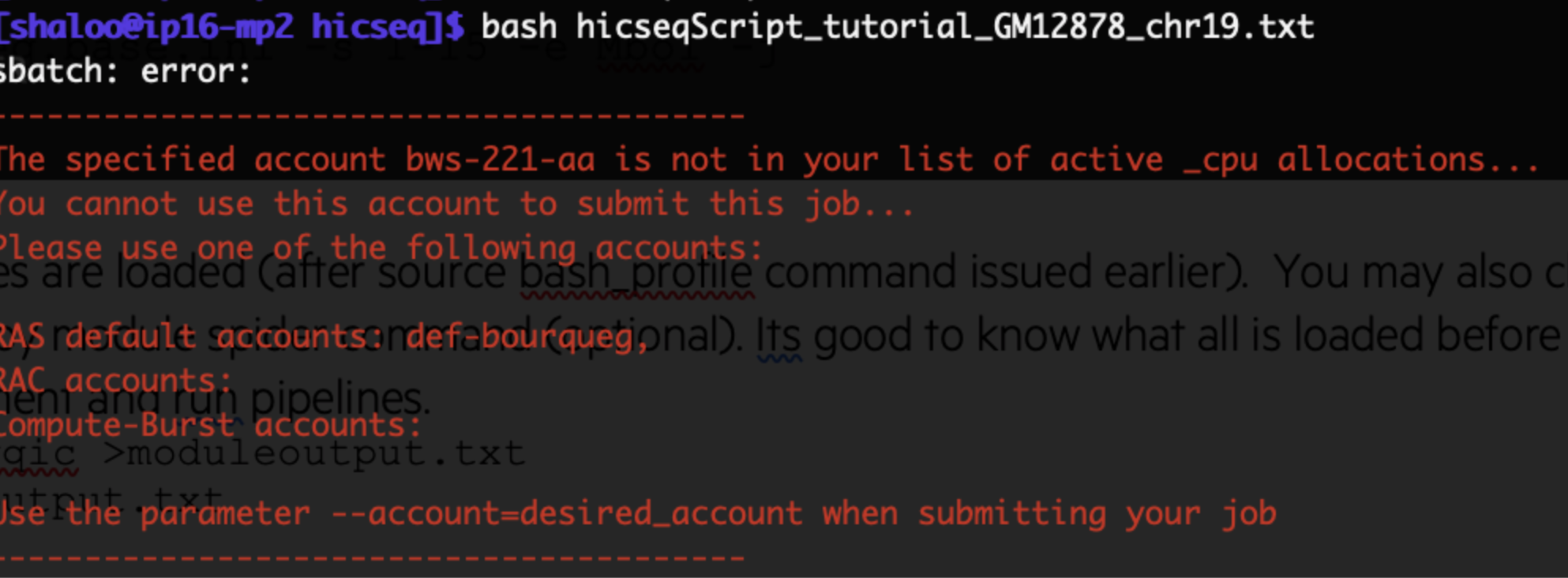
Figure: Error encountered if RAP_ID not set or set incorrectly
Fix
Make sure you have updated your .bashrc file as directed in 3. GenPipes Environment Setup. Once you set up the correct RAP_ID when you run the bash commands for your pipeline, they all go through and get scheduled depending on the scheduler (default or as as specified by -j option in pipeline command)
Runtime Failure: Job fails on worker nodes
When you issue the pipeline commands, the jobs fail to run on worker nodes.
Fix
The most common reason for this failure is not setting up the .bashrc with mugqic modules. See details on accessing GenPipes deployed on the Digital Research Alliance of Canada (DRAC), formerly Compute Canada, servers. See 3. GenPipes Environment Setup.
For other types of GenPipes deployments, Deployment Options, make sure you have closely followed the Before Running GenPipes before actually issuing GenPipes pipeline run commands.
Check: GenPipes Google Group for discussion on the latest release.