Analyzing Results
This document guides you in analyzing GenPipes pipelines results. It assumes you understand GenPipes basics and have used a pipeline (see Using GenPipes).
Viewing Results
By default, pipeline output is saved in the launch directory. You can specify a different
output directory using -o or --output-dir flags when launching the pipeline.
user@machine:~$ genpipes rnaseq -o /PATH/TO/OUTPUT ... (other options) -g genpipes_cmd_list.sh
user@machine:~$ bash genpipes_cmd_list.sh
To generate the report after a successful run, rerun the pipeline command with the --report flag.
Job Output
Most pipelines produce reports in HTML format. These are located in the job_output
For each pipeline step, the logs are stored in a corresponding sub-directory.
You can identify the top-level log file for the pipeline by its name. It follows the following naming convention:
job_output/[PIPELINE]_job_list_[YEAR-MM-DDTHH.MM.SS]
where,
[PIPELINE]: Name of the pipeline
[YEAR-MM-DDTHH.MM.SS]: The date and time of pipeline launch
This top-level job_list output file contains the status of each pipeline job run and the sub-jobs.
Warning
This job_list file is available only when you run the pipelines deployed on DRAC servers for these job schedulers:
PBS
Slurm
This feature is not supported if you run the GenPipes pipeline in a container.
Reports
In the latest GenPipes release v6.x, generating reports has changed.
Earlier, there were two different scripts for generating reports when using PBS or Slurm scheduler.
With version 6.x it is now a single command for both schedulers, log_report.
The log_report command returns the status of each job. In addition to the detailed report, it also outputs a summary file with the information about:
jobs that completed successfully
jobs that failed
jobs that are still active/inactive.
You can save the reports as .csv or .tsv files and open them in Excel on your laptop.
Use the following command to generate the tab-delimited report:
user@machine:~$ genpipes tools log_report --tsv log.out job_output/{PIPELINE}_job_list_{DATE}T{TIME}
For example:
user@machine:~$ genpipes tools log_report --tsv log.out job_output/DnaSeq_job_list_2018-06-26T12.54.27
Use the log_report.pl script to generate the tab-delimited report for Abacus:
user@machine:~$ genpipes tools log_report.pl job_output/{PIPELINE}_job_list_{DATE}T{TIME}
Example
user@machine:~$ genpipes tools log_report.pl job_output/RnaSeq_job_list_2025-09-22T10.05.27 --tsv log.out
Use the log_report.py script to generate the html report for running Slurm Scheduler on the Digital Research Alliance of Canada (DRAC), formerly Compute Canada (CCDB) servers:
user@machine:~$ genpipes tools log_report.py job_output/{PIPELINE}_job_list_{DATE}T{TIME} --tsv log.out
Example
user@machine:~$ genpipes tools log_report.py job_output/DnaSeq_job_list_2025-10-26T12.54.27 --tsv log.out
Warning
By default, the script log_report.py provides less detailed output that the new log_report.pl script in
version 6.x.
Use the –tsv option for detailed output with log_report.py.
Exit Codes
The exit code indicates status for each pipeline job run.
For example,
0: Pipeline run was successful without any issues
271: Insufficient RAM allocated to the job causing job failure
-11: Job was prematurely killed as it exceeded the allocated ‘wall-time`` due to insufficient compute resources allocation
Note
For every GenPipes Pipeline run, output is created in the default or specified location.
However, please note that what is actually written in the output location may vary significantly across different pipelines. See Pipelines Reference for specifics.
Error Logs
When a pipeline job is launched, GenPipes creates a job_output folder. All the logs and errors for the run are stored in this output
folder. Logs corresponding to each pipeline step are stored in a corresponding sub directory. If an error occurs, check the job_output folder for the specific log of the failed step and see what what printed last before it shut down. This usually helps to understand and pinpoint the real cause.
When a job finishes successfully, it will create a file with the extension .done.
See GenPipes Error Logs for details.
Visualizing Output
GenPipes output results vary a lot depending upon each specific pipeline and the way it is configured to run. Also, the way results are analyzed is also dependent on the final objective of the analysis.
For example, in case of visualizations, the results have to be imported to R or Python or some alternative visualization package.
Tools such as Integrative Genomics Viewer (IGV - Integrative Genomics Viewer), Genome Browser Gateway and several others are utilized for visualization of results. These tools vary from pipeline to pipeline.
Figure below demonstrates one such tool used for RNA Sequencing Analysis.
Figure: Genome Browser Gateway
The best way for new users and beginners to begin to explore the results is to look at the interactive MultiQC reports. Most pipelines support this and generate an html report that is saved under the report directory.
The MultiQC report that is automatically generated when running the pipeline summarizes the most important results in the pipeline, while providing tables or plots per sample. More advanced users can use the output files used as input to MultiQC to generate their own visualizations or further analyze results using their own methods.
As mentioned earlier, visualization of results varies from pipeline to pipeline. As a reference, you can see RNA Sequencing Analysis Visualization of results.
Figure below shows how the data is displayed once the alignment files are opened on IGV.
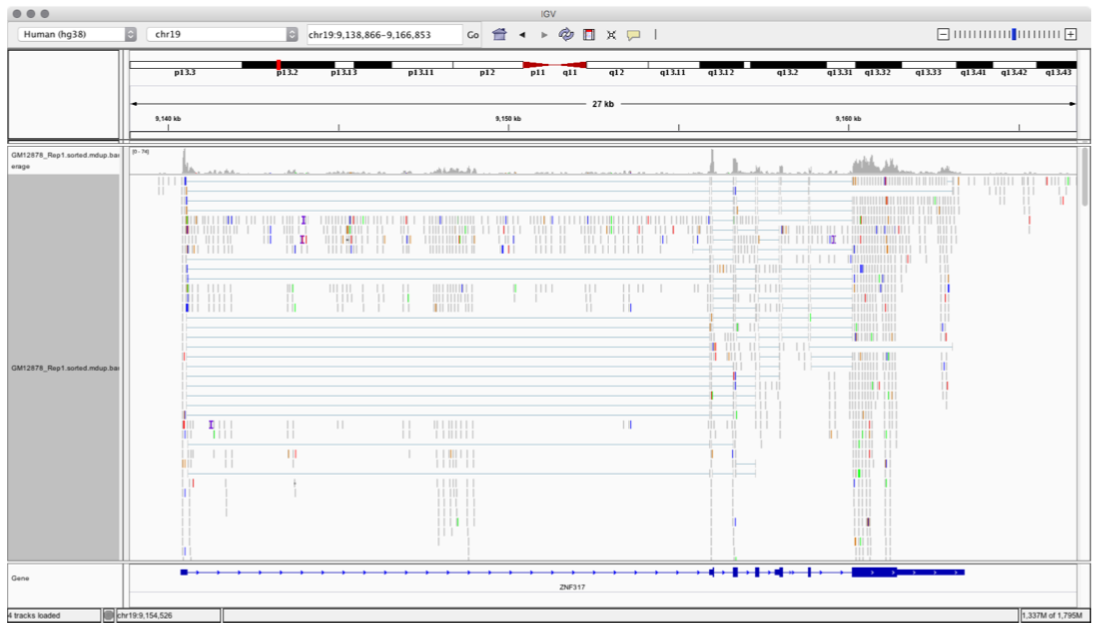
Figure: Data Alignment visualizer using IGV Tool
Relaunching Pipeline Run
If a GenPipes pipeline run fails for any reason, you can recreate the commands and relaunch the pipeline run.
When recreating the commands, GenPipes can detect job steps that have completed successfully and will not rerun those specific steps. That being said, unless you understand why a job failed in the first instance and fix it, relaunched jobs might fail with the same error.
Deleting Temporary Files
GenPipes stores some temporary files that are useful to shorten potential the reruns. To delete all these files, you can run the GenPipes command with --clean flag. This will delete a lot of files that were marked by GenPipe developers as “removable”. If you are interested in referring to these temporary files later, avoid the –clean command.
Tracking Run Parameters for Publications
In order to keep track of all parameters used, GenPipes creates a final .config.trace.ini file each time it is run. It is a good idea to keep a copy of that file in order to keep track of software versions used when publishing your paper or publication.